Embed Size (px)
Citation preview

24. ISOTOPIC COMPOSITION OF INTERSTITIAL FLUIDS AND ORIGIN OF METHANE INSLOPE SEDIMENT OF THE MIDDLE AMERICA TRENCH, DEEP SEA
DRILLING PROJECT LEG 841
George E. Claypool and Charles N. Threlkeld, U.S. Geological Survey, DenverPaul N. Mankiewicz, Global Geochemistry Corporation, Canoga Park
Michael A. Arthur, University of Rhode Islandand
Thomas F. Anderson, University of Illinois2
ABSTRACT
C H 4 and CO 2 species in pore fluids from slope sediments off Guatemala show extreme 13C-enrichment (δ13C of- 4 1 and +38‰, respectively) compared with the typical degree of 13C-enrichment in pore fluids of DSDP sediments(δ1 3C of - 60 and + lO‰). These unusual isotopic compositions are believed to result from microbial decomposition oforganic matter, and possibly from additional isotopic fractionation associated with the formation of gas hydrates. Inaddition to the isotopic fractionation displayed by CH 4 and CO2, the pore water exhibits a systematic increase in δ 1 8 θwith decrease in chlorinity. As against seawater δ 1 8 θ values of 0 and chlorinity of 19‰, the water collected from decom-posed gas hydrate from Hole 570 had a δ 1 8 θ of + 3.0‰ and chlorinity of 9.5‰. The isotopic compositions of pore-flu-id constituents change gradually with depth in Hole 568 and discontinuously with depth in Hole 570.
INTRODUCTION
Previous drilling on DSDP Legs 66 and 67 demon-strated the presence of gas-charged sediments and gashydrates on the continental slope of Middle America.The sampling and analytical programs on these earlierlegs, however, were not specifically designed for the studyof gassy sediments. In addition, the presence of gas hy-drate and the possibility of free gas beneath the gas hy-drate zone prevented achievement of some of the drill-ing objectives. Subsequently, Leg 84 was scheduled todrill in the same area as Leg 67. Drilling objectives forLeg 84 were selected in areas where gas hydrates werebelieved not to be a problem, or where they could bereached at depths shallower than the base of the gas hy-drate zone. In addition, one site was selected for thespecific study of gas hydrates, with specialized equip-ment to be deployed. This is the second site selected forintensive study of gas hydrates, the first being Site 533on Leg 76 (Kvenvolden and Barnard, 1983).
On Leg 84, Site 568 was selected for intensive studyof gas hydrates. Interstitial fluid samples were collectedat closely spaced depth intervals. The pressure core bar-rel was used in an attempt to sample gas hydrates. Read-ily observable gas hydrate was recovered in Hole 568,and an unexpected layer of relatively pure gas hydrate, 3to 4 m thick, was cored in Hole 570, with about 1 m ofgas hydrate recovered. Minor gas hydrate occurrenceswere also observed in Holes 565, and possibly in Holes566 and 569 (Kvenvolden and McDonald, this volume).
von Huene, R., Aubouin, J., et al., Init. Repts. DSDP, 84: Washington (U.S. Govt.Printing Office).
2 Addresses: (Claypool and Threlkeld) U.S. Geological Survey, MS 977, Box 25046, Den-ver, CO 80225; (Mankiewicz) Global Geochemistry Corp., Canoga Park, CA; (Arthur) Univ.of Rhode Island, Kingston, Rl; (Anderson) Univ. of Illinois, Champaign-Urbana, IL.
In this chapter, we present the results of chemical andisotopic analyses of the interstitial fluids (gas and water)from selected Holes on Leg 84. Also, we discuss the dia-genetic processes responsible for the gas and possible ef-fects of gas hydrate stability and occurrence on the op-eration of these diagenetic processes.
SAMPLES AND METHODS
Sediment gas and interstitial water were sampled according to stan-dard DSDP procedures (Gealy and Dubois, 1971; Manheim, 1966).Gases were analyzed by the procedures of Claypool et al. (1980) andSchoell (1980), with modifications. Water samples were analyzed bythe techniques of Presley and Claypool (1971) and Epstein and May-eda (1953). 1 3 C/ 1 2 C, 1 8 O/ 1 6 O, and D/H were measured by standardtechniques on CO 2 and H 2 gas; the results are expressed in the deltanotation (δ, ‰ = {[Rsamp,e/Rstandard] ~ 1} I0 3, where 7? = 1 3 C/ 1 2 C,1 8 O/ 1 6 O, D/H) relative to the standards PeeDee belemnite (PDB) forcarbon and standard mean ocean water (SMOW) for hydrogen andoxygen.
RESULTS
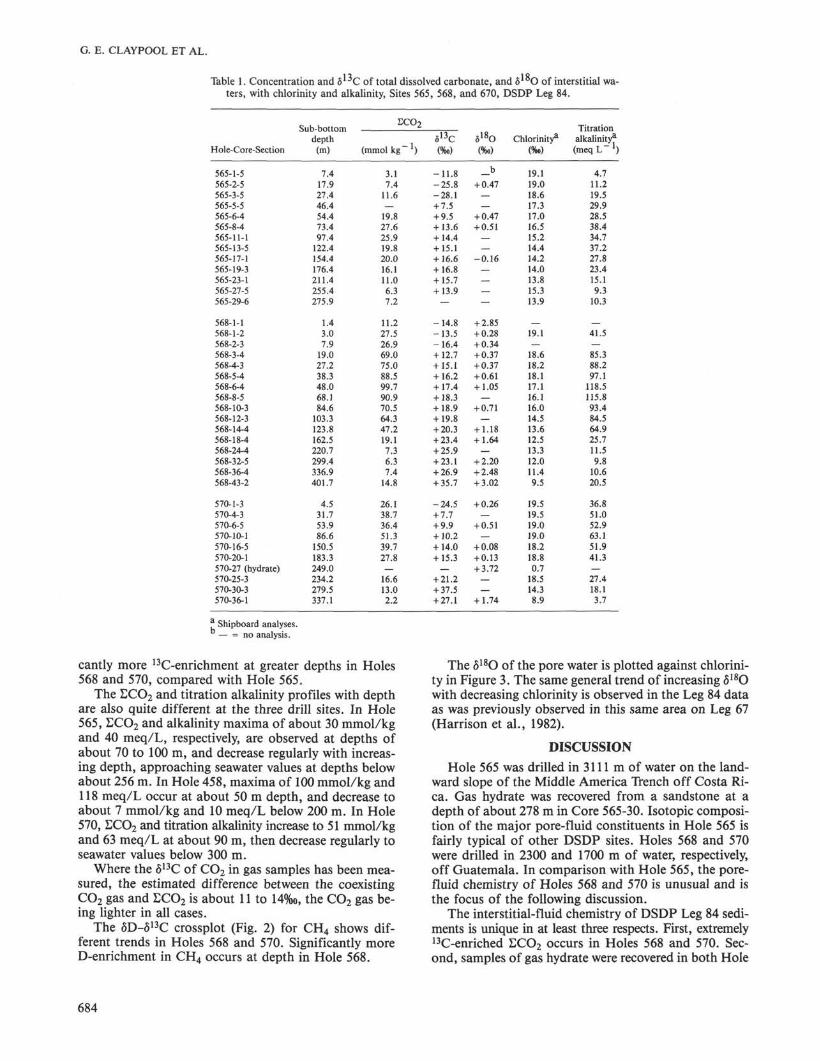
Pore-water samples from three holes (565, 568, 570)are listed by depth of burial in Table 1. The concentra-tion and δ1 3C of total dissolved carbonate (£CO2) andthe δ 1 8 θ of the water are reported, along with the ship-board determinations of chlorinity and titration alkalin-ity.
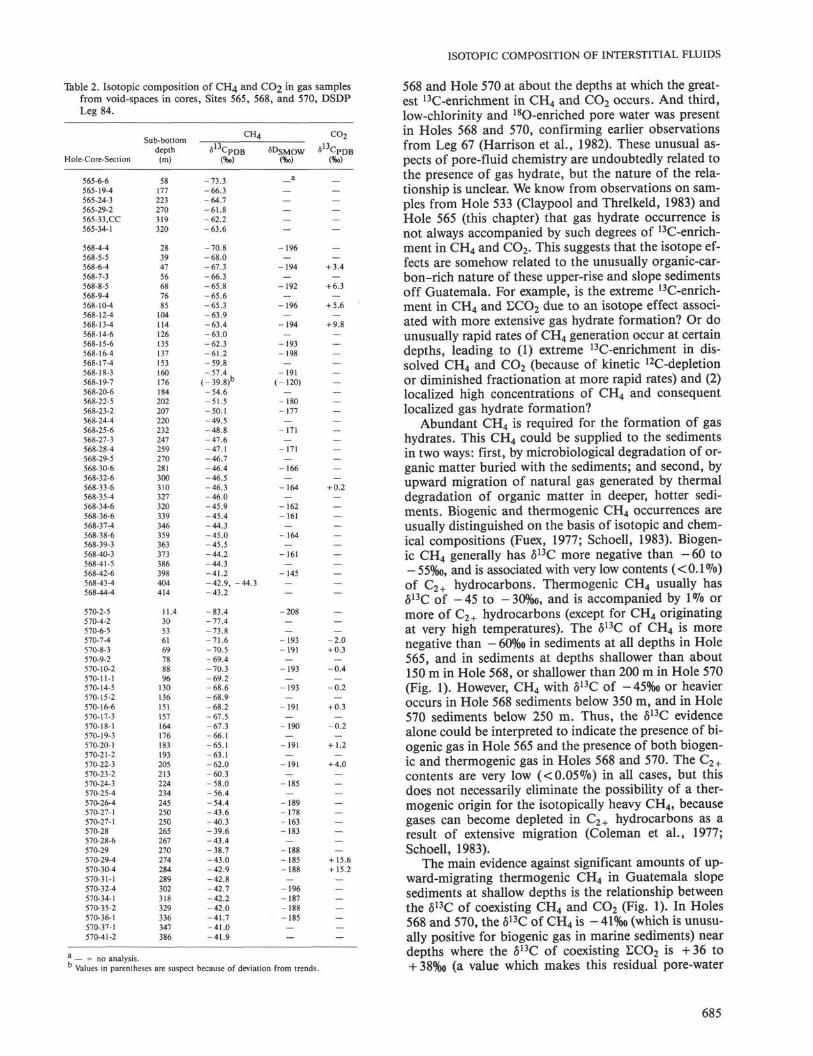
The chemical and isotopic compositions of gas sam-ples from Holes 565, 568, and 570 are given by depth ofburial in Table 2. The δ1 3C of CH 4 is given for each sam-ple. On about half of the samples, δD of the CH 4 wasmeasured, and for fewer samples, δ1 3C of the CO 2 wasalso measured.
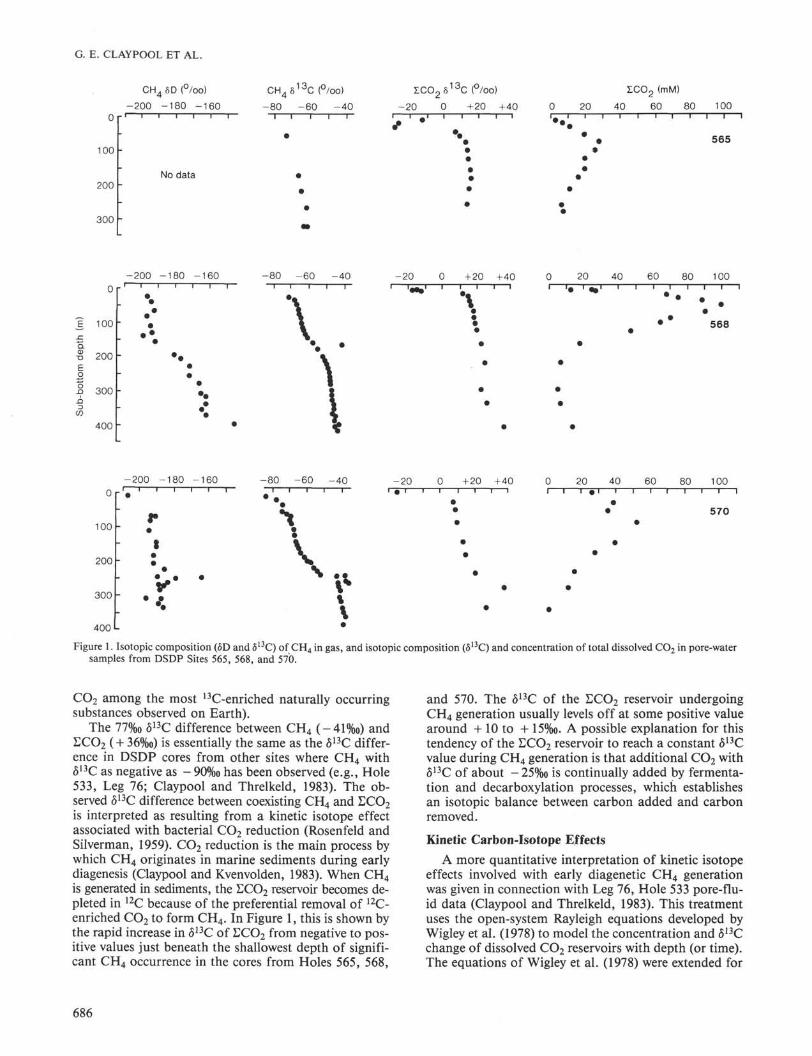
Selected data from Tables 1 and 2 are plotted in Fig-ure 1. The carbon isotopic compositions of CH 4 and to-tal dissolved CO 2 (£CO2) in the interstitial water showsimilar trends at each of the three sites, but with signifi-
683
G. E. CLAYPOOL ET AL.
-i Λ to
Table 1. Concentration and δ 1 J C of total dissolved carbonate, and δ l o θ of interstitial wa-ters, with chlorinity and alkalinity, Sites 565, 568, and 670, DSDP Leg 84.
Hole-Core-Section
565-1-5565-2-5565-3-5565-5-5565-6-4565-8-4565-11-1565-13-5565-17-1565-19-3565-23-1565-27-5565-29-6
568-1-1568-1-2568-2-3568-3-4568-4-3568-5-4568-6-4568-8-5568-10-3568-12-3568-14-4568-18-4568-24-4568-32-5568-36-4568-43-2
570-1-3570-4-3570-6-5570-10-1570-16-5570-20-1570-27 (hydrate)570-25-3570-30-3570-36-1
Sub-bottomdepth
(m)
7.417.927.446.454.473.497.4
122.4154.4176.4211.4255.4275.9
1.43.07.9
19.027.238.348.068.184.6
103.3123.8162.5220.7299.4336.9401.7
4.531.753.986.6
150.5183.3249.0234.2279.5337.1
ECO 2
(mmol kg )
3.17.4
11.6—
19.827.625.919.820.016.111.06.37.2
11.227.526.969.075.088.599.790.970.564.347.219.17.36.37.4
14.8
26.138.736.451.339.727.8—
16.613.02.2
δ 1 3 C
(‰)
-11.8-25.8-28.1+ 7.5+ 9.5+ 13.6+ 14.4+ 15.1+ 16.6+ 16.8+ 15.7+ 13.9
—
-14.8-13.5-16.4+ 12.7+ 15.1+ 16.2+ 17.4+ 18.3+ 18.9+ 19.8+ 20.3+ 23.4+ 25.9+ 23.1+ 26.9+ 35.7
-24.5+ 7.7+ 9.9+ 10.2+ 14.0+ 15.3
—+ 21.2+ 37.5+ 27.1
δ 1 8 θ
(‰)
_ b
+ 0.47——
+ 0.47+ 0.51
-0.16——_
—
+ 2.85+ 0.28+ 0.34+ 0.37+ 0.37+ 0.61+ 1.05
+ 0.71_
+ 1.18+ 1.64
_+ 2.20+ 2.48+ 3.02
+ 0.26
+ 0.51—
+ 0.08+ 0.13+ 3.72
+ 1.74
Chlorinity3
(‰)
19.119.018.617.317.016.515.214.414.214.013.815.313.9
19.1_
18.618.218.117.116.116.014.513.612.513.312.011.49.5
19.519.519.019.018.218.80.7
18.514.38.9
Titrationalkalinity*
( m e q L ~ 1 )
4.711.219.529.928.538.434.737.227.823.415.19.3
10.3
41.5_85.388.297.1
118.5115.893.484.564.925.711.59.8
10.620.5
36.851.052.963.151.941.3—
27.418.13.7
** Shipboard analyses.— = no analysis.
cantly more 13C-enrichment at greater depths in Holes568 and 570, compared with Hole 565.
The ECO2 and titration alkalinity profiles with depthare also quite different at the three drill sites. In Hole565, Σ C O 2 and alkalinity maxima of about 30 mmol/kgand 40 meq/L, respectively, are observed at depths ofabout 70 to 100 m, and decrease regularly with increas-ing depth, approaching seawater values at depths belowabout 256 m. In Hole 458, maxima of 100 mmol/kg and118 meq/L occur at about 50 m depth, and decrease toabout 7 mmol/kg and 10 meq/L below 200 m. In Hole570, Σ C O 2 and titration alkalinity increase to 51 mmol/kgand 63 meq/L at about 90 m, then decrease regularly toseawater values below 300 m.
Where the δ13C of CO2 in gas samples has been mea-sured, the estimated difference between the coexistingCO2 gas and Σ C O 2 is about 11 to 14%o, the CO2 gas be-ing lighter in all cases.
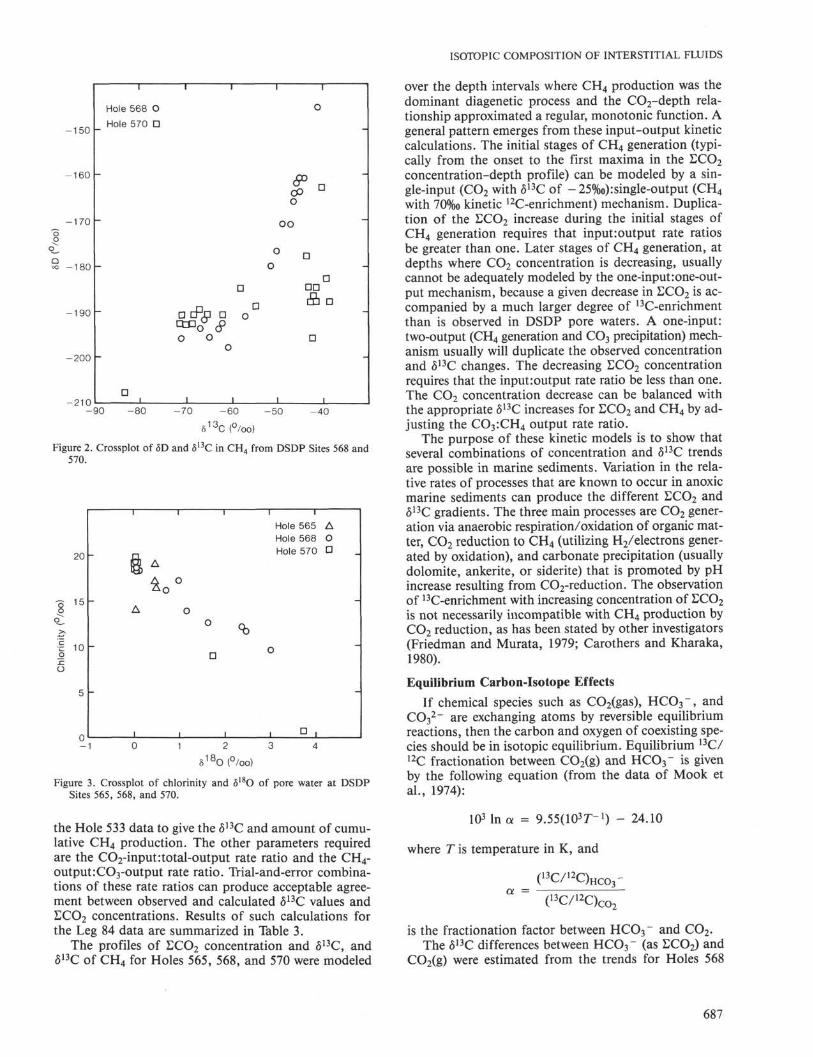
The δD-δ13C crossplot (Fig. 2) for CH4 shows dif-ferent trends in Holes 568 and 570. Significantly moreD-enrichment in CH4 occurs at depth in Hole 568.
The δ 1 8 θ of the pore water is plotted against chlorini-ty in Figure 3. The same general trend of increasing δ 1 8 θwith decreasing chlorinity is observed in the Leg 84 dataas was previously observed in this same area on Leg 67(Harrison et al., 1982).
DISCUSSION
Hole 565 was drilled in 3111 m of water on the land-ward slope of the Middle America Trench off Costa Ri-ca. Gas hydrate was recovered from a sandstone at adepth of about 278 m in Core 565-30. Isotopic composi-tion of the major pore-fluid constituents in Hole 565 isfairly typical of other DSDP sites. Holes 568 and 570were drilled in 2300 and 1700 m of water, respectively,off Guatemala. In comparison with Hole 565, the pore-fluid chemistry of Holes 568 and 570 is unusual and isthe focus of the following discussion.
The interstitial-fluid chemistry of DSDP Leg 84 sedi-ments is unique in at least three respects. First, extremely13C-enriched Σ C O 2 occurs in Holes 568 and 570. Sec-ond, samples of gas hydrate were recovered in both Hole
684
ISOTOPIC COMPOSITION OF INTERSTITIAL FLUIDS
Table 2. Isotopic composition of CH4 and CO2 m S a s samplesfrom void-spaces in cores, Sites 565, 568, and 570, DSDPLeg 84.
Hole-Core-Section
565-6-6565-19-4565-24-3565-29-2565-33,CC565-34-1
568-4-4568-5-5568-6-4568-7-3568-8-5568-9-4568-10-4568-12-4568-13-4568-14-6568-15-6568-16-4568-17-4568-18-3568-19-7568-20-6568-22-5568-23-2568-24-4568-25-6568-27-3568-28-4568-29-5568-30-6568-32-6568-33-6568-35-4568-34-6568-36-6568-37-4568-38-6568-39-3568-40-3568-41-5568-42-6568-43-4568-44-4
570-2-5570-4-2570-6-5570-7-4570-8-3570-9-2570-10-2570-11-1570-14-5570-15-2570-16-6570-17-3570-18-1570-19-3570-20-1570-21-2570-22-3570-23-2570-24-3570-25-4570-26-4570-27-1570-27-1570-28570-28-6570-29570-29-4570-30-4
570-31-1570-32-4570-34-1570-35-2570-36-1570-37-1570-41-2
Sub-bottomdepth
(m)
58177
223270319320
28394756687685
1041141261351371531601761842022072202322472592702813003!0327320339346359363373386398404414
11.430536169788896
130136151157164176183193205213224234245250250265267270274284289302318329336347386
C H 4
δ 1 3 C P D B
(‰)
-73.3-66 .3-64.7-61.8-62.2-63.6
-70.8-68 .0-67 .3-66 .3-65.8-65 .6- 6 5 . 3-63.9-63 .4-63 .0-62.3-61.2-59.8
-57.4(-39.8)D
-54.6-51.5-50.1-49.5-48.8-47.6-47.1-46.7-46.4-46.5-46.3-46.0-45.9-45.4-44.3-45.0-45.5-44.2-44.3-41.2-42.9, -44.3-43.2
-83.4-77.4-73.8-71.6-70.5-69.4-70.3-69.2-68.6-68.9-68.2-67.5-67.3-66.1-65.1-63.1-62.0-60.3-58.0-56.4-54.4-43.6-40.3-39.6-43.4-38.7-43.0-42.9-42.8-42.7-42.2-42.0-41.7-41.0-41.9
5 DSMOW(‰)
_ a
—___
—
-196—
-194—
-192—
-196—
-194—
-193-198
—
-191(-120)
—
-180- 177
—
-171—
- 171—
-166—
-164—
-162-161
_
-164—
-161—
-145—
—
-208——
-193-191
—
-193—
-193—
-191—
-190—
-191—
-191—
-185—
-189-178-163-183
—
-188-185-188
—
-196-187-188-185
—
—
co2
S 1 3 C P D B
(‰)
_
—————
—+ 3.4
—
+ 6.3—
+ 5.6—
+ 9.8———
———————————_—
+ 0.2——————————
—
——
-2.0+ 0.3
_
-0.4_
-0.2—
+ 0.3—
-0.2—
+ 1.2—
+ 4.0—————_——
+ 15.6+ 15.2
_—
————
—
a — = no analysis.Values in parentheses are suspect because of deviation from trends.
568 and Hole 570 at about the depths at which the great-est 13C-enrichment in CH 4 and CO 2 occurs. And third,low-chlorinity and 18O-enriched pore water was presentin Holes 568 and 570, confirming earlier observationsfrom Leg 67 (Harrison et al., 1982). These unusual as-pects of pore-fluid chemistry are undoubtedly related tothe presence of gas hydrate, but the nature of the rela-tionship is unclear. We know from observations on sam-ples from Hole 533 (Claypool and Threlkeld, 1983) andHole 565 (this chapter) that gas hydrate occurrence isnot always accompanied by such degrees of 13C-enrich-ment in CH 4 and CO 2 . This suggests that the isotope ef-fects are somehow related to the unusually organic-car-bon-rich nature of these upper-rise and slope sedimentsoff Guatemala. For example, is the extreme 13C-enrich-ment in CH 4 and ECO2 due to an isotope effect associ-ated with more extensive gas hydrate formation? Or dounusually rapid rates of CH 4 generation occur at certaindepths, leading to (1) extreme 13C-enrichment in dis-solved CH 4 and CO 2 (because of kinetic 12C-depletionor diminished fractionation at more rapid rates) and (2)localized high concentrations of CH 4 and consequentlocalized gas hydrate formation?
Abundant CH 4 is required for the formation of gashydrates. This CH 4 could be supplied to the sedimentsin two ways: first, by microbiological degradation of or-ganic matter buried with the sediments; and second, byupward migration of natural gas generated by thermaldegradation of organic matter in deeper, hotter sedi-ments. Biogenic and thermogenic CH 4 occurrences areusually distinguished on the basis of isotopic and chem-ical compositions (Fuex, 1977; Schoell, 1983). Biogen-ic CH 4 generally has δ1 3C more negative than - 60 to- 55%o, and is associated with very low contents (<0.1%)of C 2 + hydrocarbons. Thermogenic CH 4 usually hasδ1 3C of - 4 5 to -30‰, and is accompanied by 1% ormore of C 2 + hydrocarbons (except for CH 4 originatingat very high temperatures). The δ1 3C of CH 4 is morenegative than - 60‰ in sediments at all depths in Hole565, and in sediments at depths shallower than about150 m in Hole 568, or shallower than 200 m in Hole 570(Fig. 1). However, CH 4 with δ1 3C of -45%o or heavieroccurs in Hole 568 sediments below 350 m, and in Hole570 sediments below 250 m. Thus, the δ1 3C evidencealone could be interpreted to indicate the presence of bi-ogenic gas in Hole 565 and the presence of both biogen-ic and thermogenic gas in Holes 568 and 570. The C 2 +
contents are very low (<0.05%) in all cases, but thisdoes not necessarily eliminate the possibility of a ther-mogenic origin for the isotopically heavy CH 4, becausegases can become depleted in C 2 + hydrocarbons as aresult of extensive migration (Coleman et al., 1977;Schoell, 1983).
The main evidence against significant amounts of up-ward-migrating thermogenic CH 4 in Guatemala slopesediments at shallow depths is the relationship betweenthe δ1 3C of coexisting CH 4 and CO 2 (Fig. 1). In Holes568 and 570, the δ 1 3C of CH 4 is - 41%o (which is unusu-ally positive for biogenic gas in marine sediments) neardepths where the δ1 3C of coexisting ECO2 is +36 to+ 38‰ (a value which makes this residual pore-water
685
G. E. CLAYPOOL ET AL.
CH 4 δD (°/oo)
-200 -180 -160
CH 4 δ 1 3 C (°/oo)
- 8 0 -60 - 4 0
ICOgδ^C (°/oo)
- 2 0 +20 +40 20
I C O 2 (mM)
40 60 80 100O r '
100
200
300
' •565
No data
-200 -180 -160 -80 -60 -40 - 2 0 +20 +40Or
ε 100
200
300
400 \
' \
0 20 40 60 80 100
5 6 8
-200 -180 -160 -80 -60 -40
O r
100
200
300
400
•
-20 0 +20 +40 0 20 40 60 80 100
5 7 0
Figure 1. Isotopic composition (δD and δ13C) of C H 4 in gas, and isotopic composition (δ13C) and concentration of total dissolved CO 2 in pore-watersamples from DSDP Sites 565, 568, and 570.
CO 2 among the most 13C-enriched naturally occurringsubstances observed on Earth).
The 77%0 δ1 3C difference between CH 4 (-41‰) andΣ C O 2 ( + 36%o) is essentially the same as the δ1 3C differ-ence in DSDP cores from other sites where CH 4 withδ1 3C as negative as -90‰ has been observed (e.g., Hole533, Leg 76; Claypool and Threlkeld, 1983). The ob-served <513C difference between coexisting CH 4 and Σ C O 2
is interpreted as resulting from a kinetic isotope effectassociated with bacterial CO 2 reduction (Rosenfeld andSilverman, 1959). CO 2 reduction is the main process bywhich CH 4 originates in marine sediments during earlydiagenesis (Claypool and Kvenvolden, 1983). When CH 4
is generated in sediments, the ECO2 reservoir becomes de-pleted in 1 2C because of the preferential removal of 1 2C-enriched CO 2 to form CH 4 . In Figure 1, this is shown bythe rapid increase in δ1 3C of Σ C O 2 from negative to pos-itive values just beneath the shallowest depth of signifi-cant CH 4 occurrence in the cores from Holes 565, 568,
and 570. The δ1 3C of the ECO2 reservoir undergoingCH 4 generation usually levels off at some positive valuearound + 10 to + 15%o. A possible explanation for thistendency of the LCO2 reservoir to reach a constant δ1 3Cvalue during CH 4 generation is that additional CO2 withδ1 3C of about -25%o is continually added by fermenta-tion and decarboxylation processes, which establishesan isotopic balance between carbon added and carbonremoved.
Kinetic Carbon-Isotope Effects
A more quantitative interpretation of kinetic isotopeeffects involved with early diagenetic CH 4 generationwas given in connection with Leg 76, Hole 533 pore-flu-id data (Claypool and Threlkeld, 1983). This treatmentuses the open-system Rayleigh equations developed byWigley et al. (1978) to model the concentration and δ1 3Cchange of dissolved CO 2 reservoirs with depth (or time).The equations of Wigley et al. (1978) were extended for
686
ISOTOPIC COMPOSITION OF INTERSTITIAL FLUIDS
-150
-160
-170
-180
-190
-200
-210
Hole 568 O
Hole 570 D
O
OO
DDD
<P °
-90 -80 -70 -60 -50 -40
δ C ( /θθ)
Figure 2. Crossplot of δD and δ1 3C in CH 4 from DSDP Sites 568 and570.
20
15
c 10
o
Hole 565Hole 568Hole 570
ΔO
- 1
δ 1 8 θ
Figure 3. Crossplot of chlorinity and δ 1 8 θ of pore water at DSDPSites 565, 568, and 570.
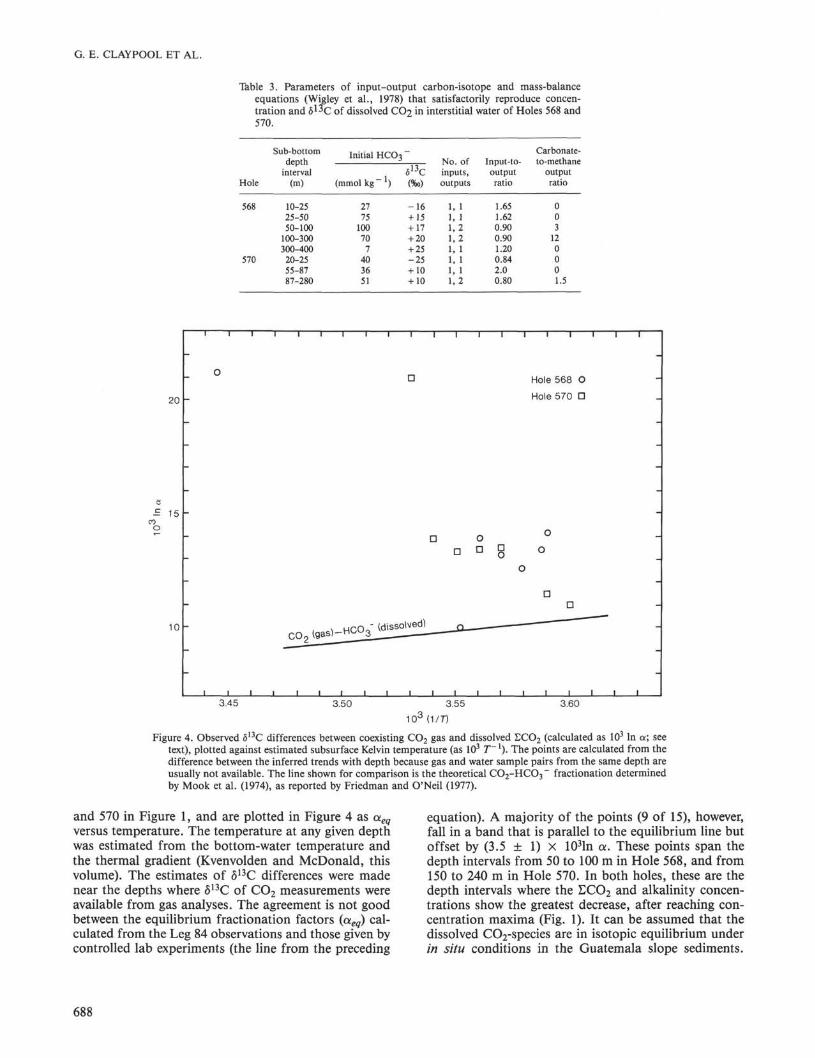
the Hole 533 data to give the δ1 3C and amount of cumu-lative CH 4 production. The other parameters requiredare the CO2-input: total-output rate ratio and the CH4-output:CO3-output rate ratio. Trial-and-error combina-tions of these rate ratios can produce acceptable agree-ment between observed and calculated δ1 3C values andΣ C O 2 concentrations. Results of such calculations forthe Leg 84 data are summarized in Table 3.
The profiles of ECO2 concentration and δ1 3C, andδ1 3C of CH 4 for Holes 565, 568, and 570 were modeled
over the depth intervals where CH 4 production was thedominant diagenetic process and the CO2-depth rela-tionship approximated a regular, monotonic function. Ageneral pattern emerges from these input-output kineticcalculations. The initial stages of CH 4 generation (typi-cally from the onset to the first maxima in the Σ C O 2
concentration-depth profile) can be modeled by a sin-gle-input (CO2 with δ1 3C of -25%o):single-output (CH4
with 7O%o kinetic 12C-enrichment) mechanism. Duplica-tion of the Σ C O 2 increase during the initial stages ofCH 4 generation requires that input:output rate ratiosbe greater than one. Later stages of CH 4 generation, atdepths where CO 2 concentration is decreasing, usuallycannot be adequately modeled by the one-input:one-out-put mechanism, because a given decrease in Σ C O 2 is ac-companied by a much larger degree of 13C-enrichmentthan is observed in DSDP pore waters. A one-input:two-output (CH4 generation and CO3 precipitation) mech-anism usually will duplicate the observed concentrationand δ1 3C changes. The decreasing LCO2 concentrationrequires that the input:output rate ratio be less than one.The CO 2 concentration decrease can be balanced withthe appropriate δ1 3C increases for Σ C O 2 and CH 4 by ad-justing the CO 3 :CH 4 output rate ratio.
The purpose of these kinetic models is to show thatseveral combinations of concentration and δ1 3C trendsare possible in marine sediments. Variation in the rela-tive rates of processes that are known to occur in anoxicmarine sediments can produce the different Σ C O 2 andδ1 3C gradients. The three main processes are CO 2 gener-ation via anaerobic respiration/oxidation of organic mat-ter, CO 2 reduction to CH 4 (utilizing H2/electrons gener-ated by oxidation), and carbonate precipitation (usuallydolomite, ankerite, or siderite) that is promoted by pHincrease resulting from CO2-reduction. The observationof 13C-enrichment with increasing concentration of Σ C O 2
is not necessarily incompatible with CH 4 production byCO 2 reduction, as has been stated by other investigators(Friedman and Murata, 1979; Carothers and Kharaka,1980).
Equilibrium Carbon-Isotope Effects
If chemical species such as CO2(gas), HCO 3 ~, andCO 3
2~ are exchanging atoms by reversible equilibriumreactions, then the carbon and oxygen of coexisting spe-cies should be in isotopic equilibrium. Equilibrium 1 3 C/1 2C fractionation between CO2(g) and H C O 3 " is givenby the following equation (from the data of Mook etal., 1974):
1 0 3 l n α = 9.55(103r-!) - 24.10
where T is temperature in K, and
a —( 1 3 C / 1 2 Q c θ 2
is the fractionation factor between HCO 3 and CO 2.The δ1 3C differences between HCO 3~ (as Σ C O 2 ) and
CO2(g) were estimated from the trends for Holes 568
687
G. E. CLAYPOOL ET AL.
Table 3. Parameters of input-output carbon-isotope and mass-balanceequations (Wigley et al., 1978) that satisfactorily reproduce concen-tration and δ ^ c of dissolved CO2 m interstitial water of Holes 568 and570.
Hole
568
570
Sub-bottomdepth
interval(m)
10-2525-5050-100
100-300300-40020-2555-8787-280
Initial HCO3
(mmol kg )
2775
10070
7403651
-
δ 1 3 C(‰)
- 1 6+ 15+ 17+ 20+ 25- 2 5+ 10+ 10
No ofinputs,outputs
1, 11, 11, 21, 21, 11, 11, 11, 2
outputratio
1.651.620.900.901.200.842.00.80
Carbonate-to-methane
outputratio
003
120001.5
20
£ 15
10
Hole 568 O
Hole 570 D
DO
D α o
CO
3.45 3.50 3.55 3.60
10 3 (1/T)
Figure 4. Observed δ 1 3C differences between coexisting CO 2 gas and dissolved ECO2 (calculated as I03 In α; seetext), plotted against estimated subsurface Kelvin temperature (as I03 T~'). The points are calculated from thedifference between the inferred trends with depth because gas and water sample pairs from the same depth areusually not available. The line shown for comparison is the theoretical CO 2 -HCO 3 ~ fractionation determinedby Mook et al. (1974), as reported by Friedman and O'Neil (1977).
and 570 in Figure 1, and are plotted in Figure 4 as aeq
versus temperature. The temperature at any given depthwas estimated from the bottom-water temperature andthe thermal gradient (Kvenvolden and McDonald, thisvolume). The estimates of δ13C differences were madenear the depths where δ13C of CO2 measurements wereavailable from gas analyses. The agreement is not goodbetween the equilibrium fractionation factors (aeq) cal-culated from the Leg 84 observations and those given bycontrolled lab experiments (the line from the preceding
equation). A majority of the points (9 of 15), however,fall in a band that is parallel to the equilibrium line butoffset by (3.5 ± 1) × 103ln a. These points span thedepth intervals from 50 to 100 m in Hole 568, and from150 to 240 m in Hole 570. In both holes, these are thedepth intervals where the LCO2 and alkalinity concen-trations show the greatest decrease, after reaching con-centration maxima (Fig. 1). It can be assumed that thedissolved CO2-species are in isotopic equilibrium underin situ conditions in the Guatemala slope sediments.
688
ISOTOPIC COMPOSITION OF INTERSTITIAL FLUIDS
Therefore, the disagreement shown in Figure 4 must bedue to various imperfections in the sampling and analyt-ical procedures.
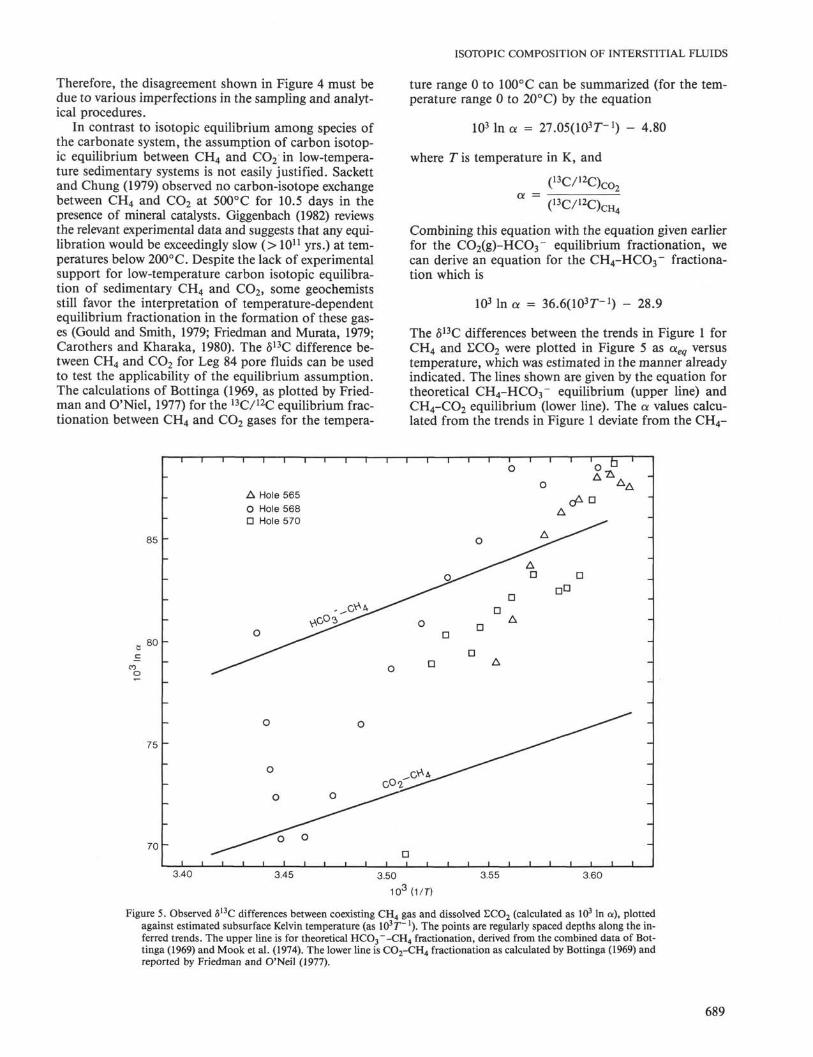
In contrast to isotopic equilibrium among species ofthe carbonate system, the assumption of carbon isotop-ic equilibrium between CH4 and CO2 in low-tempera-ture sedimentary systems is not easily justified. Sackettand Chung (1979) observed no carbon-isotope exchangebetween CH4 and CO2 at 500°C for 10.5 days in thepresence of mineral catalysts. Giggenbach (1982) reviewsthe relevant experimental data and suggests that any equi-libration would be exceedingly slow (> I011 yrs.) at tem-peratures below 200° C. Despite the lack of experimentalsupport for low-temperature carbon isotopic equilibra-tion of sedimentary CH4 and CO2, some geochemistsstill favor the interpretation of temperature-dependentequilibrium fractionation in the formation of these gas-es (Gould and Smith, 1979; Friedman and Murata, 1979;Carothers and Kharaka, 1980). The δ13C difference be-tween CH4 and CO2 for Leg 84 pore fluids can be usedto test the applicability of the equilibrium assumption.The calculations of Bottinga (1969, as plotted by Fried-man and O'Niel, 1977) for the 13C/12C equilibrium frac-tionation between CH4 and CO2 gases for the tempera-
ture range 0 to 100°C can be summarized (for the tem-perature range 0 to 20°C) by the equation
I03 In a = 27.05(103r-1) - 4.80
where T is temperature in K, and
( 1 3C/ 1 2C) c θ 2
a =
Combining this equation with the equation given earlierfor the CO2(g)-HCO3" equilibrium fractionation, wecan derive an equation for the CH4-HCO3~ fractiona-tion which is
lOMnα = 36.6(103r-1) - 28.9
The δ13C differences between the trends in Figure 1 forCH4 and ECO2 were plotted in Figure 5 as aeq versustemperature, which was estimated in the manner alreadyindicated. The lines shown are given by the equation fortheoretical CH4-HCO3~ equilibrium (upper line) andCH4-CO2 equilibrium (lower line). The a values calcu-lated from the trends in Figure 1 deviate from the CH4-
85
80
75
70
i T^ i
Δ Hole 565O Hole 568D Hole 570
3.40 3.45 3.50
103 (1/7)
3.55 3.60
Figure 5. Observed δ 1 3C differences between coexisting C H 4 gas and dissolved ECO2 (calculated as I0 3 In α), plottedagainst estimated subsurface Kelvin temperature (as 103T~ l). The points are regularly spaced depths along the in-ferred trends. The upper line is for theoretical HCO 3 ~-CH 4 fractionation, derived from the combined data of Bot-tinga (1969) and Mook et al. (1974). The lower line is CO 2 -CH 4 fractionation as calculated by Bottinga (1969) andreported by Friedman and O'Neil (1977).
689

G. E. CLAYPOOL ET AL.
HCO 3~ equilibrium iine by about + 4 to -20‰. More-over, the detailed trends in the δ1 3C difference withincreasing depth are significantly different from that pre-dicted by temperature-dependent equilibrium. The cal-culated values are in only general agreement with whatis predicted by equilibrium considerations. The kineticapproach to interpreting CH 4 -CO 2 δ1 3C differences ismore useful because it incorporates consideration ofamounts of reactants and products, and the relative ratesof the biological processes.
Gas-Hydrate Isotope Effects
The correlation between increasing δ 1 8 θ and decreas-ing chlorinity or salinity of pore water in sediments onthe continental slope off Guatemala was first observedon Leg 67 (Harrison et al., 1982; Hesse and Harrison,1981). The pore-water δ 1 8 θ analyses reported in Table 1and shown in Figure 3 for samples from Holes 568 and570 confirm the Leg 67 observations. The water mole-cules that form the solid clathrate hydrate are known toexclude salts and concentrate 1 8O, relative to the coexist-ing liquid water (Davidson et al., 1983). The fractiona-tion factor, a = ( 1 8O/ 1 6O) s o l i d/( 1 8O/ 1 6O) l i q u i d, is believedto be about the same as that for the ice-water equilib-rium (O'Neil, 1968), or about 1.003. As shown in Table 1,pore-water samples with normal chlorinities have δ 1 8 θvalues in the range of 0.3 to O.5%o, whereas water col-lected from decomposed gas hydrate from Core 27 ofHole 570 has δ 1 8 θ of 3.72‰. If 0.5 and 3.7%0 are takenas the δ 1 8 θ values of the liquid and solid H 2O, a valueof α = 1.0032 is obtained.
The question of what happens to the salts and 1 8O-depleted H 2 O excluded during gas hydrate formationhas not been resolved, but loss to overlying seawater oradjacent sediments seems to be required. An earlier in-terpretation (Hesse and Harrison, 1981) was that the gashydrate probably is in contact with pore water of nor-mal salinity and δ 1 8 θ . In this view, the observed "fresh-ening" and 18O-enrichment is due to decomposition ofgas hydrates during sampling and dilution of the porewater with water from the hydrate. The jagged depth-profiles for δ 1 8 θ and chlorinity at Site 533 of Leg 76support this interpretation (Jenden and Geiskes, 1983).However, results obtained with the in situ pore-watersampler in Hole 568 do not support this interpretationfor the Guatemala slope sediments. The chlorinities ofthe in situ pore-water samples showed about the sameCl~-depletion as squeezed pore-water samples from sim-ilar depths. It should also be noted that pore water inHole 570 does not show the regular changes in chlorini-ty and δ 1 8 θ with depth. Only the water from decom-posed gas hydrate at 249 m and the deepest samples (at280 and 337 m) shows significant departure from seawa-ter/pore-water chlorinity and δ 1 8 θ .
Is the 18O-enrichment in H 2O of gas hydrates accom-panied by similar 1 3C- and D-enrichment in the CH 4 ofgas hydrate? There is reason to expect that there wouldbe some isotopic effect (Trofimuk et al., 1974), but thepossible magnitude of such an effect is difficult to eval-uate. CH 4 hydrate is not easy to study experimentally,and the conditions required for its formation in the lab-oratory (vigorous stirring) make meaningful observation
of possible isotopic effects difficult. No obvious isotop-ic effect has been observed during controlled decompo-sition of CH 4 hydrate. Indirect evidence that gas hy-drates are not the major cause of 13C-fractionation isprovided by the fact that similar patterns of δ1 3C changewith depth are observed in regions where gas hydratesare not stable, such as the Cariaco Trench (Claypooland Kaplan, 1974).
SUMMARY AND CONCLUSIONS
The pore-fluid samples from sediments in Holes 568and 570 have extremely 13C-enriched early diagenetic CH 4
and CO2 ( -41 and + 38%o, respectively). The sedimentsalso have very high contents of organic matter (3-4%organic carbon), and the best-developed marine gas-hy-drate occurrences observed to date. The abundant or-ganic matter should have supported prolonged and vig-orous microbiological CH 4 generation, with consequentextreme 13C-depletion in the SCO 2 reservoir from whichthe CH 4 was formed, and more extensive developmentof gas hydrates. Alternatively, methane generation couldhave resulted in the degree of 12C-enrichment that is typ-ical for deep-sea sediments, and the development of gashydrate may have superimposed additional fractionationand resulted in the observed extreme 13C-enrichment. Res-olution of these alternative interpretations will have toawait an improved understanding of the relevant diage-netic processes.
Isotopic compositions of other pore-fluid constitu-ents (δD of CH 4 , δ 1 8 θ of H2O) in continental-rise sedi-ments of the Guatemala margin also exhibit systematictrends. The trends of isotopic composition with depth atthe two sites differ significantly where gas hydrates ei-ther are massively developed at intermediate depths (Hole570), or are disseminated and sufficiently developed topermit recovery only in the deepest part of the sectionpenetrated (Hole 568). These different isotope-depth gra-dients may represent two different diagenetic styles orsituations: one (Hole 570) in which the gradients are pre-dominantly diffusion-controlled and gas hydrate devel-opment is concentrated at some preferred zone (e.g., be-neath an unconformity), and another (Hole 568) in whichthere is progressive development of gas hydrate with in-creasing depth, and the gradients are predominantlythose of steady-state diagenesis.
The pore-fluid data from DSDP Leg 84 provide abun-dant material for developing hypotheses regarding earlydiagenetic processes of the decomposition of organicmatter in continental-margin sediments. Geochemical in-vestigations to test these hypotheses should play a majorrole in planning for future deep-sea drilling.
ACKNOWLEDGMENTS
This work was funded in part by the Morgantown Energy Tech-nology Center under USGS-USDOE interagency agreement no. DE-AI21-83MC20422, and by Gas Research Institute Contract No. 5081-360-0533 to Global Geochemistry Corporation. Walter E. Dean andZvi Sofer reviewed the manuscript, and D. R. Malone typed it.
REFERENCES
Bottinga, Y., 1969. Calculated fractionation factors for carbon andhydrogen isotope exchange in the system calcite-CO2-graphite-methane-hydrogen and water vapor. Geochim. Cosmochim. Acta,33:49-64.
690

ISOTOPIC COMPOSITION OF INTERSTITIAL FLUIDS
Carothers, W. W., and Kharaka, Y. K., 1980. Stable carbon isotopesof HCO3~ in oil-field waters—implications for the origin of CO2.Geochim. Cosmochim. Acta, 44:323-332.
Claypool, G. E., and Kaplan, I. R., 1974. The origin and distributionof methane in marine sediments. In Kaplan, I. R. (Ed.), NaturalGases in Marine Sediments: New York (Plenum), pp. 94-129.
Claypool, G. E., and Kvenvolden, K. A., 1983. Methane and otherhydrocarbon gases in marine sediment. Ann. Rev. Earth Planet.Sci., 11:299-328.
Claypool, G. E., and Threlkeld, C. N., 1983. Anoxic diagenesis andmethane generation in sediments of the Blake Outer Ridge, DeepSea Drilling Project Site 533, Leg 76. In Sheridan, R. E., Grad-stein, F. M., et al., Init. Repts. DSDP, 76: Washington (U.S. Govt.Printing Office), 391-402.
Claypool, G. E., Threlkeld, C. N., and Magoon, L. B., 1980. Biogen-ic and thermogenic origins of natural gas in Cook Inlet Basin,Alaska. Am. Assoc. Pet. Geol. Bull., 64:1131-1139.
Coleman, D. D., Lin, C , and Keogh, R. A., 1977. Isotopic identifica-tion of leakage gas from underground storage reservoirs—a pro-gress report: ///. State Geol. Surv. III. Petrol., 111:1-10.
Davidson, D. W., Leaist, D. G., and Hesse, R., 1983. Oxygen-18 en-richment in the water of a clathrate hydrate. Geochim. Cosmo-chim. Acta, 47:2293-2295.
Epstein, S., and Mayeda, T., 1953. Variation of oxygen-18 contentof waters from natural sources. Geochim. Cosmochim. Acta, 4:213-224.
Friedman, I., and Murata, K. J., 1979. Origin of dolomite in MioceneMonterey Shale and related formations in the Temblor Range, Cal-ifornia. Geochim. Cosmochim. Acta, 43:1357-1365.
Friedman, I., and O'Neil, J. R., 1977. Compilation of stable isotopefractionation factors of geochemical interest. Data of Geochemis-try (6th ed.), Geol. Surv. Prof. Pap. U.S., 440-KK:l-12.
Fuex, A. N., 1977. The use of stable carbon isotopes in hydrocarbonexploration. J. Geochem. Explor., 7:155-188.
Gealy, E. L., and Dubois, R., 1971. Shipboard geochemical analysis,Leg 7, Glomar Challenger. In Winterer, E. L., Riedel, W. R., etal., Init. Repts. DSDP, 7, Pt. 2: Washington (U.S. Govt. PrintingOffice), 863-870.
Giggenbach, W. F., 1982. Carbon-13 exchange between CO2 and CH4
under geothermal conditions. Geochim. Cosmochim. Acta, 46:159-165.
Gould, K. W., and Smith, J. W., 1979. The genesis and isotopic com-position of carbonates associated with some Permian Australiancoals. Chern. Geol., 24:137-150.
Harrison, W. E., Hesse, R., and Geiskes, J. M., 1982. Relationshipbetween sedimentary facies and interstitial water chemistry of slope,trench, and Cocos Plate sites from the Middle America Trench
transect, active margin off Guatemala, DSDP Leg 67. In Aubouin,J., von Huene, R., et al., Init. Repts. DSDP, 67: Washington (U.S.Govt. Printing Office), 603-614.
Hesse, R., and Harrison, W. E., 1981. Gas hydrates (clathrates) caus-ing pore-water freshening and oxygen isotope fractionation in deep-water sedimentary sections of terrigenous continental margins.Earth Planet. Sci. Lett., 55:453-462.
Jenden, P. D., and Geiskes, J. M., 1983. Chemical and isotopic com-position of interstitial water from Deep Sea Drilling Project Sites533 and 534. In Sheridan, R. E., Gradstein, F. M., et al., In-it. Repts. DSDP, 76: Washington (U.S. Govt. Printing Office),453-462.
Kvenvolden, K. A., and Barnard, L. A., 1983. Gas hydrates of theBlake Outer Ridge, Site 533, Deep Sea Drilling Project Leg 76. InSheridan, R. E., Gradstein, F. M., et al., Init. Repts. DSDP, 76:Washington (U.S. Govt. Printing Office) 353-366.
Manheim, F. T., 1966. A hydraulic squeezer for obtaining interstitialwater from consolidated and unconsolidated sediments. Geol. Surv.Prof. Pap. U.S., 550-C:256-261.
Mook, W. G., Bommerson, J. C , and Staverman, W. H., 1974. Car-bon isotope fractionation between dissolved bicarbonate and gas-eous carbon dioxide. Earth Planet. Sci. Lett., 22:169-176.
O'Neil, J. R., 1968. Hydrogen and oxygen isotope fractionation be-tween ice and water. J. Phys. Chern., 72:3683-3684.
Presley, B. J., and Claypool, G. E., 1971. Techniques for analyzing in-terstitial water samples. Part II: Determination of total carbonateand carbon isotope ratios. In Winterer, E. L., Riedel, W. R., et al.,Init. Repts. DSDP, 1, Pt. 2: Washington (U.S. Govt. Printing Of-fice), 1756-1757.
Rosenfeld, W. D., and Silverman, S. R., 1959. Carbon isotope frac-tionation in bacterial production of methane. Science, 130:1658.
Sackett, W. M., and Chung, H. M., 1979. Experimental confirmationof the lack of carbon isotope exchange between methane and car-bon oxides at high temperatures. Geochim. Cosmochim. Acta, 43:273-276.
Schoell, M., 1980. The hydrogen and carbon isotopic composition ofmethane from natural gases of various origins. Geochim. Cosmo-chim. Acta, 44:649-661.
, 1983. Genetic characterization of natural gases. Am. As-soc. Pet. Geol. Bull., 67:2225-2238.
Trofimuk, A. A., Cherskiy, N. V. and Tsarev, V. P., 1974. Mechanismsfor fractionation of isotopes of water and gas in crustal zones ofhydrate formation. Dokl. Akad. Nauk SSSR, 215:1226-1229.
Wigley, T. M. L., Plummer, L. N., and Pearson, F. J., Jr., 1978. Masstransfer and carbon-isotope evolution in natural water systems.Geochim. Cosmochim. Acta, 42:1117-1140.
691





























