Embed Size (px)
Citation preview

15. Lecture WS 2008/09
Bioinformatics III 1
V15 Stochastic simulations of cellular signalling
Introduction into stochastic processes,
see e.g. Nico van Kampen‘s book
A random number or stochastic variable is an
object X defined by
(a) a set of possible values (called „range“, „set of states“,
„sample space“, or „phase space“) and
(b) a probability distribution over this set.
Ad (a) The set may be discrete, e.g. head or tails, or the number of molecules of a
certain component in a reacting mixture.
Or the set may be continuous in a given interval as the velocity of a Brownian
particle. Or it may be partly discrete, partly continuous.
The set of states may also be multidimensional. Then, X stands for a vector X.

15. Lecture WS 2008/09
Bioinformatics III 2
Stochastic variables
Ad (b) The probability distribution is given by a nonnegative function P(x),
0xPand normalized in the sense
1dxxP
where the integral extends over the whole range.
The probability that X has a value between x and x + dx is
dxxP

15. Lecture WS 2008/09
Bioinformatics III 3
Averages
The set of states and the probability distribution together fully define the stochastic
variable. The average or expectation value of any function f(X) defined on the
same state is dxxPxfXf
In particular,
mmX
is called the m-th moment of X, and 1 the average or mean.
212
22 XX
is called the variance, which is the square of the standard deviation .

15. Lecture WS 2008/09
Bioinformatics III 4
Addition of stochastic variables
Let X1 and X2 be two variables with joint probability density PX(x1,x2) .
The probability that Y = X1 + X2 has a value between y and y + y is
yyxxy
XY dxdxxxPyyP21
2121 ,
From this follows
111
212121
,
,
dxxyxP
dxdxxxPyxxyP
X
XY
If X1 and X2 are independent this equation becomes
111 21dxxyPxPyP XXY
Thus the probability density of the sum of two independent variables is the
convolution of their individual probability densities.

15. Lecture WS 2008/09
Bioinformatics III 5
Addition of stochastic variables
From this follows two rules concerning the moments
21 XXY
The average of the sum is the sum of the averages,
regardless of whether X1 and X2 are independent or not.
If X1 and X2 are uncorrelated,222
21 XXY

15. Lecture WS 2008/09
Bioinformatics III 6
Stochastic processes Once a stochastic variable X has been defined, an infinite number of other
stochastic variables derive from it, namely all quantities Y that are defined
as functions of X by some mapping f.
These quantities Y may be any kind of mathematical object, in particular also
functions of an additional variable t,
Such a quantity Y(t) is called a random function, or, since in most cases
t stands for time, a stochastic process.
Thus, a stochastic process is simply a function of two variables, one of which is
time t and the other a stochastic variable X.
On inserting for X one of its possible values x, an ordinary function of t obtains
called a sample function or realization of the process.
tXftYx ,
txftYx ,

15. Lecture WS 2008/09
Bioinformatics III 7
Stochastic processes It is easy to form averages, on the basis of the given probability density PX(x) of X.
E.g. dxxPtYtY Xx
A Markov process is defined as a stochastic process with the property that for any
set of n successive times, i.e. t1 < t2 < ... < tn one has
1111111111 ,,,;...;,, nnnnnnnnn tytyPtytytyP
The notation P1|n -1 means that the probability to have a particular value yn at 1 time
point tn depends on the values at n-1 previous time points.
The equality means that the conditional probability density at tn , given the value yn-1
at tn-1 is uniquely determined and, for a Markov process, is not affected by any
knowledge of the values at earlier times. P1|1 is called the transition probability.

15. Lecture WS 2008/09
Bioinformatics III 8
Markov property A Markov process is fully determined by the two functions P1|1(y1,t1) and
P1|1 (y2,t2 | y1,t1); the whole hierarchy can be constructed from them.
One obtains for instance, taking t1 < t2 < t3
Continuing this algorithm one finds successively all Pn.
This property makes Markov processes manageable, which is the reason
why they are so useful in applications.
From here one can derive the Chapman-Kolmogorov equation
223311112211111
22113321221123322113
,,,,,
,;,,,;,,;,;,
tytyPtytyPtyP
tytytyPtytyPtytytyP
2112211223311113311 ,,,,,, dytytyPtytyPtytyP
This identity must be obeyed by the transition probability of any Markov process.
The time ordering is t1 < t2 < t3 .

15. Lecture WS 2008/09
Bioinformatics III 9
Master equation
In practice, the Chapman-Kolmogorov equation is not very convenient for deriving
transition probabilities because it is a functional relation.
A more convenient version of the same equation is the Master equation.
W(y2|y1) is the transition probability per unit time from y1 to y2 .
This equation must be interpreted as follows.
Take a time t1 and a value y1, and consider the solution that is determined for t t1
by the initial condition P(y,t1) = (t – t1).
This solution is the transition probability Tt-t1 (y|y1) of the Markov process – for any
choice of t1 and y1.
',','',
dytyPyyWtyPyyWt
tyP

15. Lecture WS 2008/09
Bioinformatics III 10
Stochastic simulations of cellular signalling
Traditional computational approach to chemical/biochemical kinetics:
(a) start with a set of coupled ODEs (reaction rate equations) that describe the
time-dependent concentration of chemical species,
(b) use some integrator to calculate the concentrations as a function of time
given the rate constants and a set of initial concentrations.
Successful applications : studies of yeast cell cycle, metabolic engineering,
whole-cell scale models of metabolic pathways (E-cell), ...
Major problem: cellular processes occur in very small volumes and
frequently involve very small number of molecules.
E.g. in gene expression processes a few TF molecules may interact
with a single gene regulatory region.
E.coli cells contain on average only 10 molecules of Lac repressor.

15. Lecture WS 2008/09
Bioinformatics III 11
Include stochastic effects
(Consequence1) modeling of reactions as continuous fluxes of matter
is no longer correct.
(Consequence2) Significant stochastic fluctuations occur.
To study the stochastic effects in biochemical reactions stochastic formulations
of chemical kinetics and Monte Carlo computer simulations have been used.
Daniel Gillespie (J Comput Phys 22, 403 (1976); J Chem Phys 81, 2340 (1977))
introduced the exact Dynamic Monte Carlo (DMC) method that connects the
traditional chemical kinetics and stochastic approaches.
Assuming that the system is well mixed, the rate constants appearing in these two
methods are related.

15. Lecture WS 2008/09
Bioinformatics III 12
Dynamic Monte Carlo
In the usual implementation of DMC for kinetic simulations, each reaction is
considered as an event and each event has an associated probability of occurring.
The probability P(Ei) that a certain chemical reaction Ei takes place in a given time
interval t is proportional to an effective rate constant k and to the number of
chemical species that can take part in that event.
E.g. the probability of the first-order reaction
X Y + Z
would be k1Nx with Nx :number of species X, and
k1 : rate constant of the reaction
Similarly, the probability of the reverse second-order reaction
Y + Z X
would be k2NYNZ.
Resat et al., J.Phys.Chem. B 105, 11026 (2001)

15. Lecture WS 2008/09
Bioinformatics III 13
Dynamic Monte Carlo
As the method is a probabilistic approach based on „events“, „reactions“ included in
the DMC simulations do not have to be solely chemical reactions.
Any process that can be associated with a probability
can be included as an event in the DMC simulations.
E.g. a substrate attaching to a solid surface can initiate
a series of chemical reactions.
One can split the modelling into
- the physical events of substrate arrival,
- attaching the substrate,
- followed by the chemical reaction steps.
Resat et al., J.Phys.Chem. B 105, 11026 (2001)

15. Lecture WS 2008/09
Bioinformatics III 14
Basic outline of the direct method of Gillespie
(Step i) generate a list of the components/species and define the initial distribution
at time t = 0.
(Step ii) generate a list of possible events Ei (chemical reactions as well as
physical processes).
(Step iii) using the current component/species distribution, prepare a probability
table P(Ei) of all the events that can take place.
Compute the total probability
P(Ei) : probability of event Ei .
(Step iv) Pick two random numbers r1 and r2 [0...1] to decide
which event E will occur next and the amount of time
by which E occurs later since the most recent event.
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
)(itotEPP

15. Lecture WS 2008/09
Bioinformatics III 15
Basic outline of the direct method of Gillespie
Using the random number r1 and the probability table,
the event E is determined by finding the event that satisfies the relation
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
1
1
11
ii
itoti
EPPrEP
The second random number r2 is used to obtain the amount of time between the
reactions
2ln
1r
Ptot
As the total probability of the events changes in time, the time step between
occurring steps varies.
Steps (iii) and (iv) are repeated at each step of the simulation.
The necessary number of runs depends on the inherent noise
of the system and on the desired statistical accuracy.

15. Lecture WS 2008/09
Bioinformatics III 16
Weighted SamplingIn the commonly used MC algorithm, the Markov chain is generated using
transition probabilities (i j) that are based on the physical probability
distribution:
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
kki
ji
P
Pji
The ensemble average of any physical quantity is obtained by taking the
arithmetic average of all the n simulation runs.
The individual averages i could e.g. be time-averages over the simulation run.
This choice disfavors the transitions with low probabilities.
If the system characteristics depend on the events that happen less frequently,
then the common implementation of MC requires extremely lengthy simulations to
acquire enough statistical sampling.
n
iin 1
1

15. Lecture WS 2008/09
Bioinformatics III 17
Weighted SamplingThis statistical sampling problem can be reduced if the probability distribution is
multiplied with a weight function that adjusts the sampling probability distribution
such that the low probability parts of the sampling space are visited more often.
In the case of weighted sampling, the Markov chain is generated by using the
modified probability distribution function
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
jiYjiPjiPw
where Y is the biasing weight function.
Since the probability of the transition i j is weighted with Y(i j),
calculation of the ensemble average of a physical quantity
is obtained by computing the average of / Y.
Division of by Y effectively corrects for the bias introduced
in the sampling probability distribution.

15. Lecture WS 2008/09
Bioinformatics III 18
Probability-Weighted DMC
Probability-weighted DMC incorporates weighted sampling into DMC.
Steps (iii) and (iv) of the DMC algorithm are replaced by
(Step iii‘) Using the current component/species distribution,
prepare a probability table of all the events Ei that can take place,
(Step iv‘) define the weight factor scale and compute the inverse
probability weight table
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
EYEw 1
for all events.
Note that the stochastic simulations mentioned here use discrete numbers of
molecules, i.e. the species are produced and consumed as whole integer units.
Therefore, the weight table w(E ) must contain only integer values.

15. Lecture WS 2008/09
Bioinformatics III 19
Probability-Weighted DMC
(Step v‘) Prepare the weighted probability table
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
i
i
iw Ew
EPEP
(Step vi‘) Compute the total probability by summing the weighted probabilities of all
individual events )(
iwtotEPP
(Step vii‘) Pick two random numbers r1,r2 [0...1].
Determine which event E occurs next as before using r1.
(Step viii‘) Propagate the time as before using r2.
The speed-up achieved by the PW-DMC algorithm stems from the fact that the
reactions with large probabilities are allowed to occur in „bundles“.

15. Lecture WS 2008/09
Bioinformatics III 20
Comparison of DMC and PW-DMCDMC is essentially a method to solve the master equation that rules
how the probabilities of the configurations are related to each other
Resat et al., J.Phys.Chem. B 105, 11026 (2001)
PWPWdt
dP
W : transition probability of going from configuration to
P : probability of configuration .
Using the master equation, the statistical average X of the rate of change of the
property X can be expressed as:
,
,XXPW
dt
Xd
In PW-DMC, this relation is rearranged using the weight factor w as
,
, XXww
PW
dt
Xd
PW-DMC leaves the ensemble averages unchanged.
However, the fluctuations increase with w.

15. Lecture WS 2008/09
Bioinformatics III 21
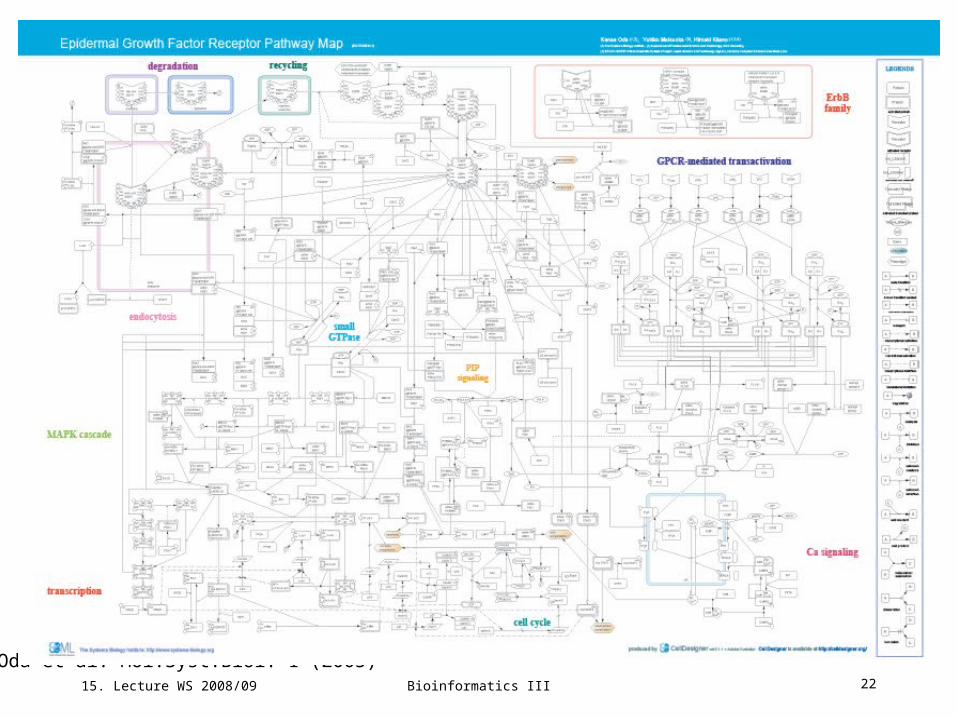
Epidermal growth factor receptor signaling pathway
The EGFR signaling pathway is one of the most important pathways that regulate
growth, survival, proliferation, and differentiation in mammalian cells.
International consortium has assembled a comprehensive pathway map including
- EGFR endocytosis followed by its degradation or recycling,
- small GTPase-mediated signal transduction such as MAPK cascade, PIP
signaling, cell cycle, and GPCR-mediated EGFR transactivation via intracellular
Ca2+ signalling.
Map includes 211 reactions and 322 species taking part in reactions.
Species: 202 proteins, 3 ions, 21 simple molecules, 73 oligomers, 7 genes, 7 RNAs.
Proteins: 122 molecules including 10 ligands, 10 receptors, 61 enzymes (including 32 kinases), 3 ion
channels, 10 transcription factors, 6 G protein subunits, 22 adaptor proteins.
Reactions: 131 state transitions, 34 transportations, 32 associations, 11 dissociations, 2 truncations.
Oda et al. Mol.Syst.Biol. 1 (2005)
15. Lecture WS 2008/09
Bioinformatics III 22
Oda et al. Mol.Syst.Biol. 1 (2005)
15. Lecture WS 2008/09
Bioinformatics III 23
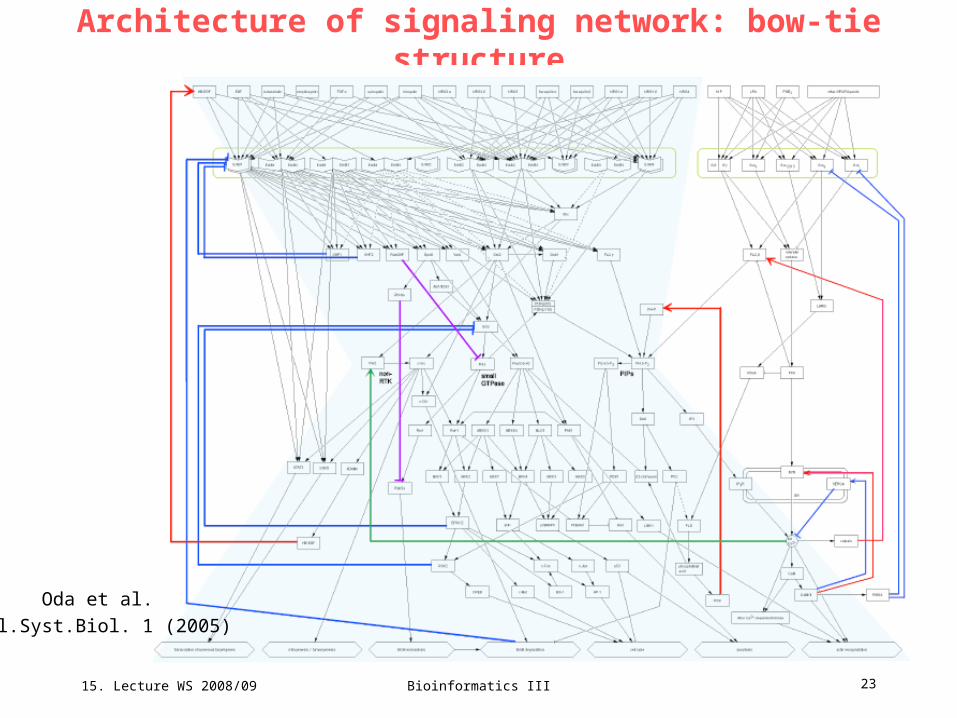
Architecture of signaling network: bow-tie structure
Oda et al.
Mol.Syst.Biol. 1 (2005)

15. Lecture WS 2008/09
Bioinformatics III 24
Network control
Several system controls define the overall behavior of the signaling network:
- 2 positive feedback loops
- Pyk2/c-Src activates ADAMs, which shed pro-HB-EGF so that the
amount of HB-EGF will be increased and enhance the signalling
- active PLC/ produces DAG which results in the cascading activation
of protein kinase C (PKC), phospholipase D, and PI5 kinase.
- 6 negative feedback loops
- inhibitory feed-forward paths
There are also a few positive and negative feedback loops that affect ErbB
pathway dynamics.
Oda et al. Mol.Syst.Biol. 1 (2005)
15. Lecture WS 2008/09
Bioinformatics III 25
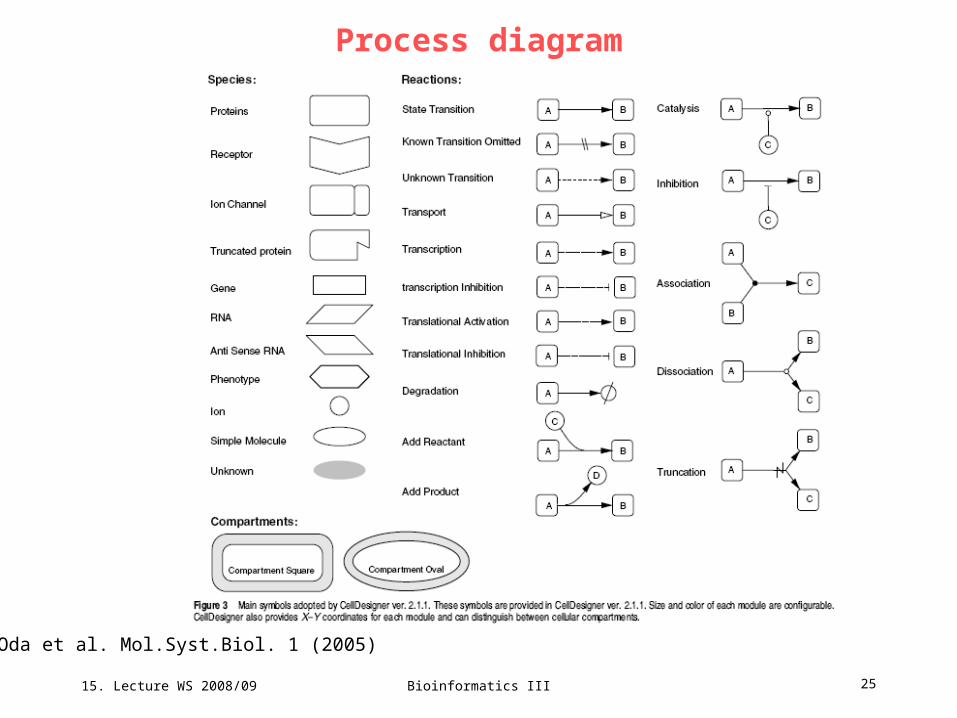
Process diagram
Oda et al. Mol.Syst.Biol. 1 (2005)

15. Lecture WS 2008/09
Bioinformatics III 26
Modification and localization of proteins
Oda et al. Mol.Syst.Biol. 1 (2005)
15. Lecture WS 2008/09
Bioinformatics III 27
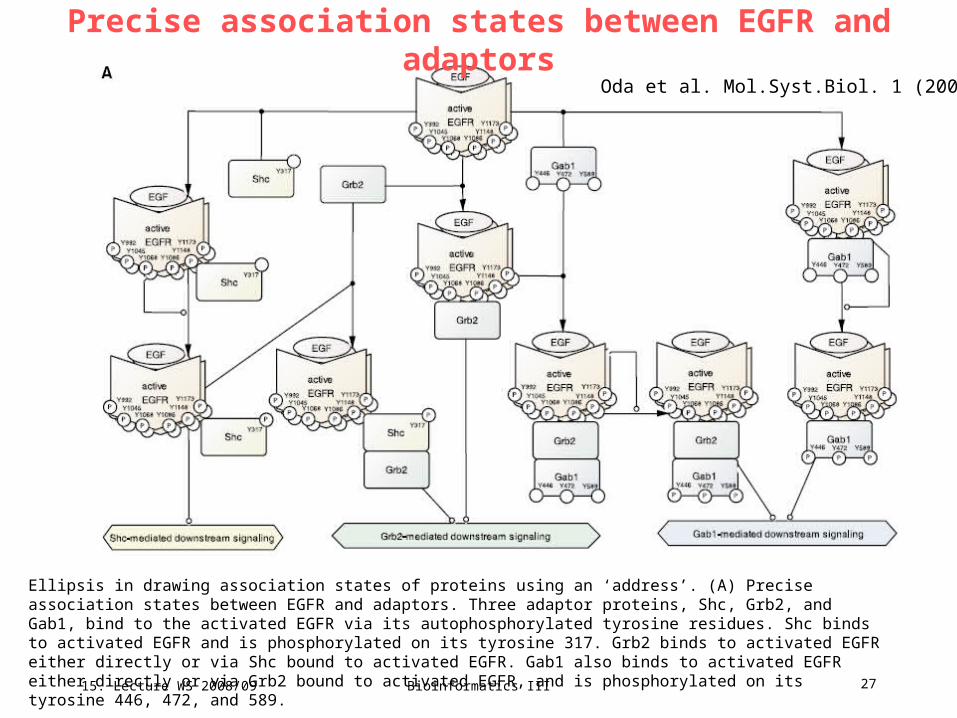
Precise association states between EGFR and adaptorsOda et al. Mol.Syst.Biol. 1 (2005)
Ellipsis in drawing association states of proteins using an ‘address’. (A) Precise association states between EGFR and adaptors. Three adaptor proteins, Shc, Grb2, and Gab1, bind to the activated EGFR via its autophosphorylated tyrosine residues. Shc binds to activated EGFR and is phosphorylated on its tyrosine 317. Grb2 binds to activated EGFR either directly or via Shc bound to activated EGFR. Gab1 also binds to activated EGFR either directly or via Grb2 bound to activated EGFR, and is phosphorylated on its tyrosine 446, 472, and 589.
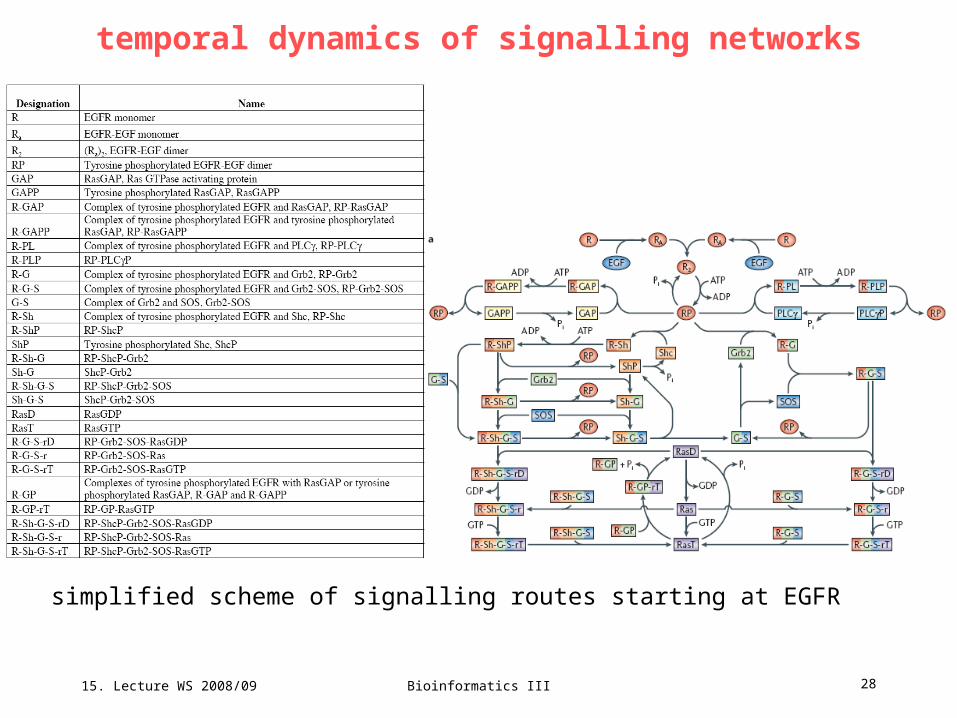
15. Lecture WS 2008/09
Bioinformatics III 28
temporal dynamics of signalling networks
simplified scheme of signalling routes starting at EGFR

15. Lecture WS 2008/09
Bioinformatics III 29
Integrated Model of Epidermal Growth Factor Receptor Trafficking and Signal Transduction
The EGF receptor can be activated by the
binding of any one of a number of different
ligands.
Each ligand stimulates a somewhat different
spectrum of biological responses.
The effect of different ligands on EGFR
activity is quite similar at a biochemical level
the mechanisms responsible for their
differential effect on cellular responses are
unkown.
After binding of any of its ligands, EGFR is
rapidly internalized by endocytosis. Haluk Resat et al.
Biophys Journal
85, 730 (2003)

15. Lecture WS 2008/09
Bioinformatics III 30
Computational modelling of EGF receptor system
(1) trafficking and ligand-induced endocytosis
(2) signaling through Ras or MAP kinases
This work combines both aspects into a single model.
Most approaches to building computational kinetic models have severe
drawbacks when representing spatially heterogenous processes on a cellular
scale.
Review: In the traditional approach, we
- formulate set of coupled ODEs (reaction rate equations) for the time-dependent
concentration of chemical species
- use integrator to propagate the concentrations as a function of time given the
rate constants and a set of initial concentrations.
Resat et al. Biophys Journal 85, 730 (2003)

15. Lecture WS 2008/09
Bioinformatics III 31
Multiple time scale problem
In Dynamic Monte Carlo, reactions are considered events that occur with certain
probabilities over set intervals of time.
The event probabilities depend on the rate constant of the reaction and on the
number of molecules participating in the reaction.
In many interesting natural problems, the time scales of the events are spread
over a large spectrum.
Therefore it is very inefficient to treat all processes at the time scale of the fastest
individual reaction.
In the EGFR signaling network,
- receptor phosphorylation after ligand binding occurs almost instantaneously
- vesicle formation or sorting to lysosomes requires many minutes.
Resat et al. Biophys Journal 85, 730 (2003)

15. Lecture WS 2008/09
Bioinformatics III 32
Solution to multiple time scale problem
Computing millions and billions non-correlated random numbers
can become a time-consuming process.
Resat et al. (2001) introduced Probability-Weighted DMC to speed-up the
simulation by factor 20 – 100.
Different processes are only tested at variant times depending on their
probabilities
very unlikely processes compute MC decision very infrequently.
Resat et al. Biophys Journal 85, 730 (2003)
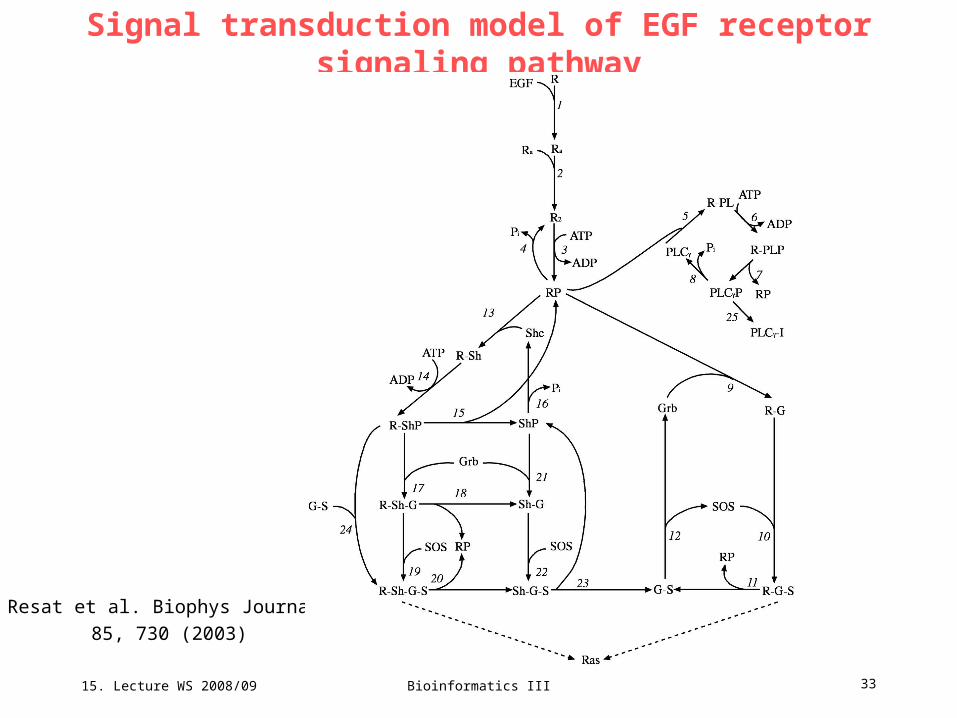
15. Lecture WS 2008/09
Bioinformatics III 33
Signal transduction model of EGF receptor signaling pathway
Resat et al. Biophys Journal
85, 730 (2003)

15. Lecture WS 2008/09
Bioinformatics III 34
Species in the EGF receptor signaling model
Resat et al. Biophys Journal
85, 730 (2003)
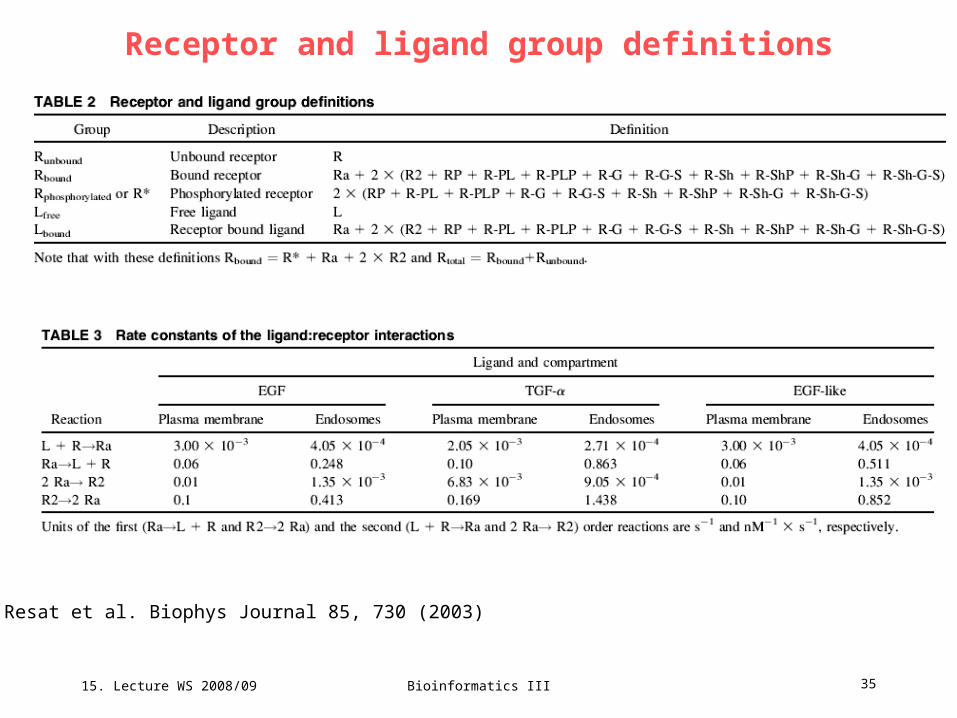
15. Lecture WS 2008/09
Bioinformatics III 35
Receptor and ligand group definitions
Resat et al. Biophys Journal 85, 730 (2003)

15. Lecture WS 2008/09
Bioinformatics III 36
Early endosome inclusion coefficients
Resat et al. Biophys Journal 85, 730 (2003)
These are adjusted to yield the experimentally determined rates of
ligand-free and ligand-bound receptor internalization.
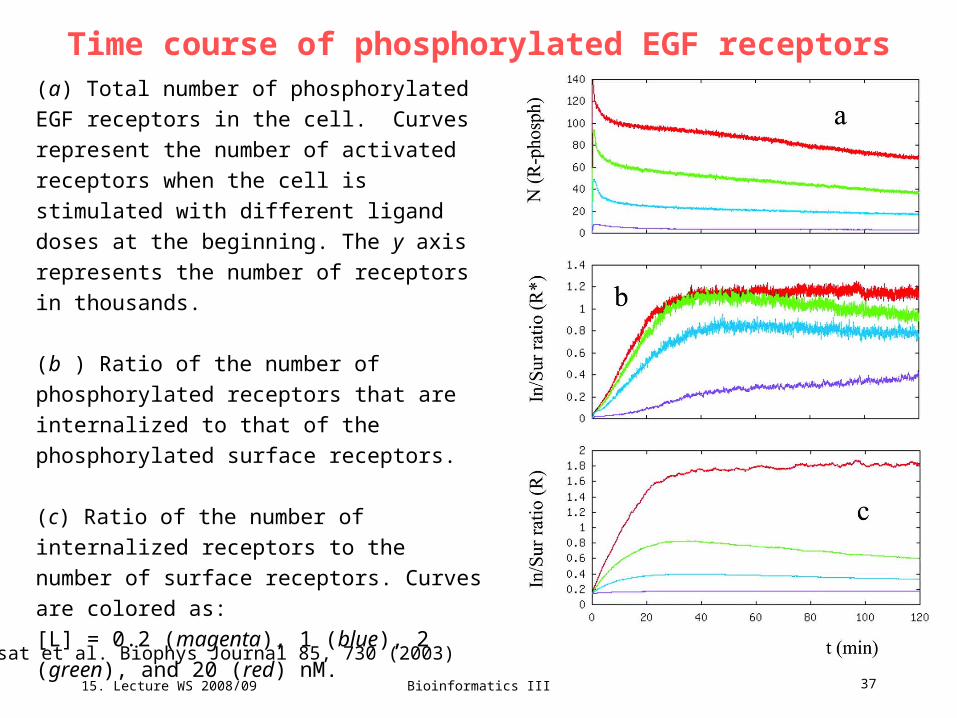
15. Lecture WS 2008/09
Bioinformatics III 37
Time course of phosphorylated EGF receptors(a) Total number of phosphorylated EGF
receptors in the cell. Curves represent the
number of activated receptors when the cell is
stimulated with different ligand doses at the
beginning. The y axis represents the number of
receptors in thousands.
(b ) Ratio of the number of phosphorylated
receptors that are internalized to that of the
phosphorylated surface receptors.
(c) Ratio of the number of internalized
receptors to the number of surface receptors.
Curves are colored as:
[L] = 0.2 (magenta), 1 (blue), 2 (green), and 20
(red) nM.
Resat et al. Biophys Journal 85, 730 (2003)
15. Lecture WS 2008/09
Bioinformatics III 38
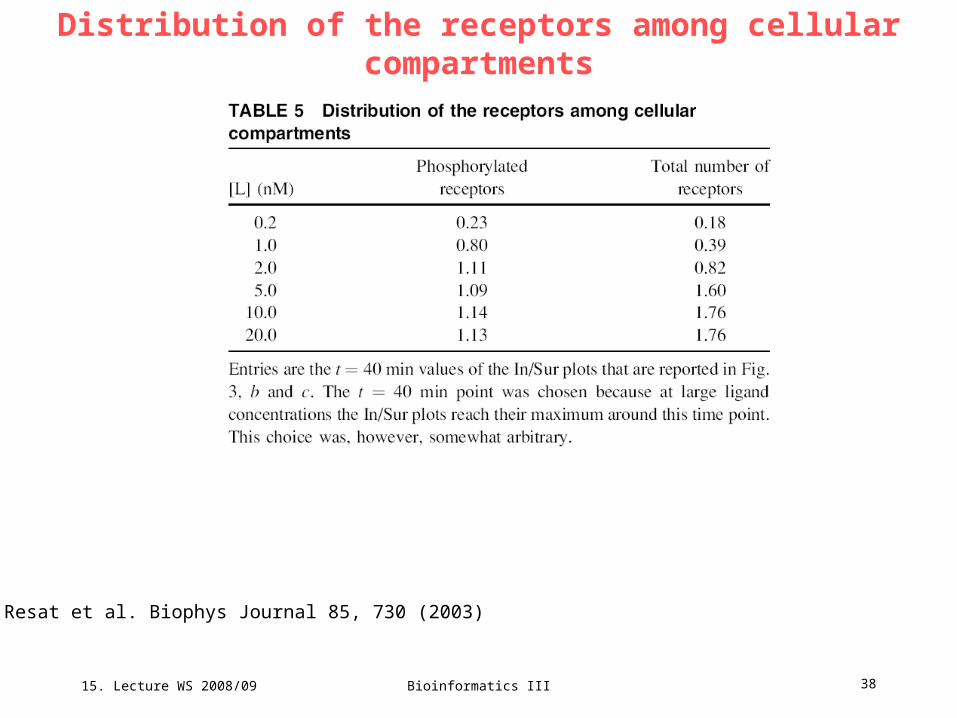
Distribution of the receptors among cellular compartments
Resat et al. Biophys Journal 85, 730 (2003)
15. Lecture WS 2008/09
Bioinformatics III 39
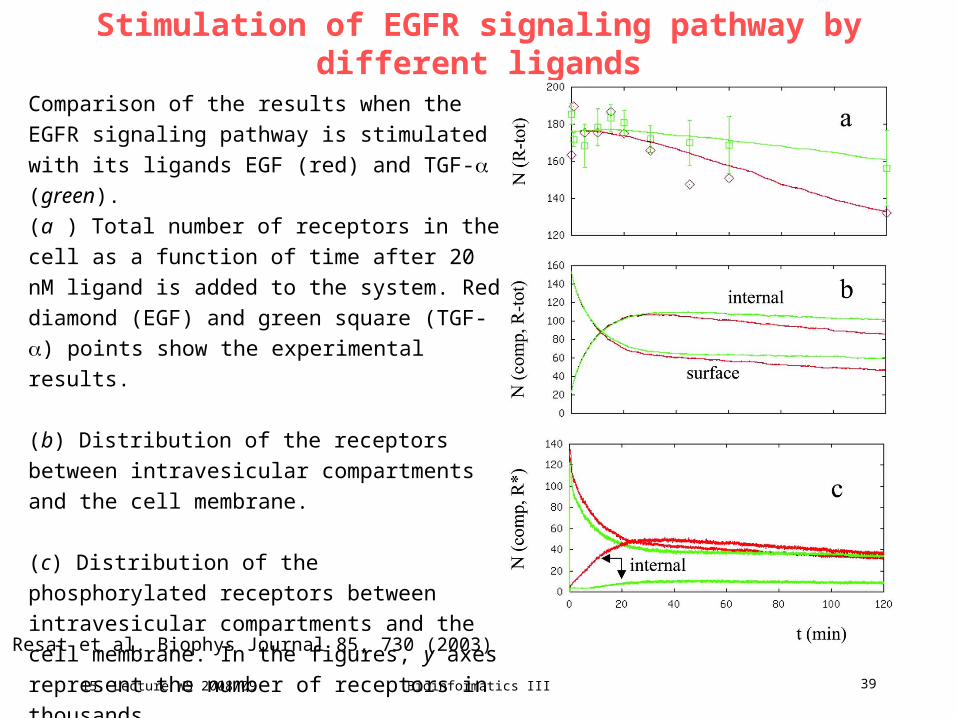
Stimulation of EGFR signaling pathway by different ligands
Comparison of the results when the EGFR
signaling pathway is stimulated with its ligands
EGF (red) and TGF- (green).
(a ) Total number of receptors in the cell as a
function of time after 20 nM ligand is added to the
system. Red diamond (EGF) and green square
(TGF-) points show the experimental results.
(b) Distribution of the receptors between
intravesicular compartments and the cell
membrane.
(c) Distribution of the phosphorylated receptors
between intravesicular compartments and the cell
membrane. In the figures, y axes represent the
number of receptors in thousands.
Resat et al. Biophys Journal 85, 730 (2003)
15. Lecture WS 2008/09
Bioinformatics III 40
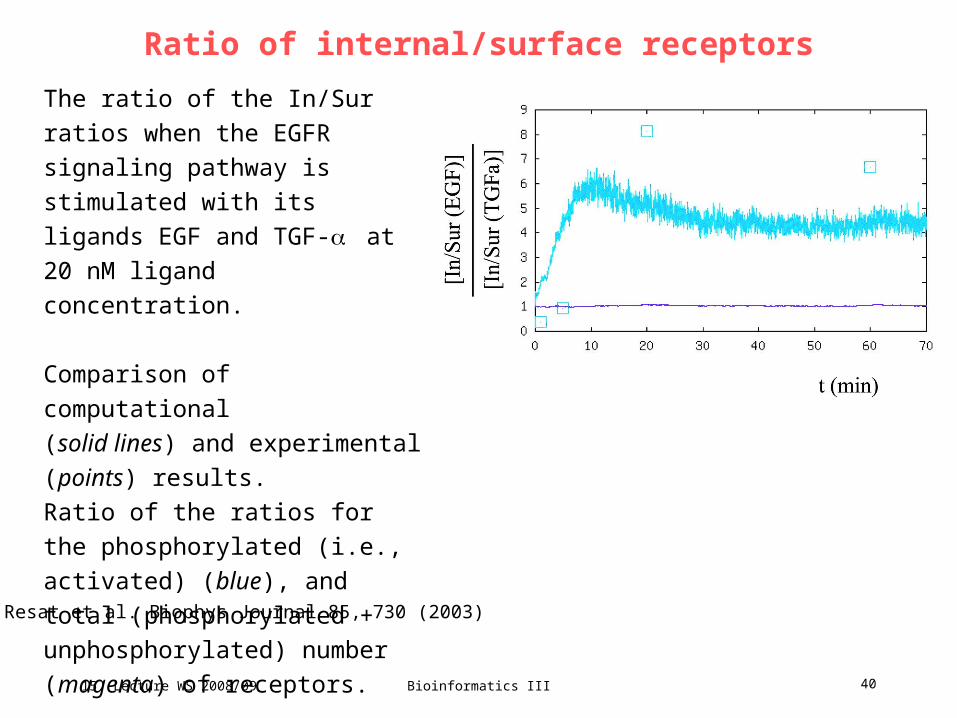
Ratio of internal/surface receptors
The ratio of the In/Sur ratios when
the EGFR signaling pathway is
stimulated with its ligands EGF and
TGF- at 20 nM ligand
concentration.
Comparison of computational
(solid lines) and experimental
(points) results.
Ratio of the ratios for the
phosphorylated (i.e., activated)
(blue), and total (phosphorylated +
unphosphorylated) number
(magenta) of receptors.
Resat et al. Biophys Journal 85, 730 (2003)

15. Lecture WS 2008/09
Bioinformatics III 41
SummaryLarge-scale simulations of the kinetics of biological signaling networks are
becoming feasible.
Here, the model of the EGFR trafficking consisted of hundreds of distinct
compartments and ca. 13.000 reactions/events that occur on a wide spatial-
temporal range.
The exact Dynamic Monte Carlo algorithm of Gillespie (1976/1977) was a
breakthrough for simulations of stochastic systems.
Problem: simulations can become very time-consuming. In particular if the
processes occur on different time scales.
Methods like the probability-weighted DMC are promising tools for studying
complex cellular systems using molecular quanta.
V16: more on stochastic dynamics simulations.





























