Embed Size (px)
Citation preview
12.4.1 Introduction
Since ancient times men have known of the uniqueproperties of an aromatic balsam, known as Styraxliquidus, extracted from Liquidambar orientalis, acommon tree throughout the Asia Minor. Thissubstance by solidifying in the presence of airallowed the hardening or waterproofing of textilesand other materials. The name styrene was given bythe German chemist Eduard Simon, who in 1839perfected a method for distilling Styrax, thusobtaining the styrene monomer (Scheirs and Priddy,2003). During his work, Simon also noted that, in thepresence of oxygen, styrene turned into a solidsubstance, which he called metastyrene. Themacromolecular character of metastyrene was thentheorised and proven by Hermann Staudinger in the1920s (Staudinger, 1932). Staudinger is alsoresponsible for its structural study, which identified arelationship between absence of stereoregularity andthe amorphous character of polystyrene (PS). Thestudies undertaken by Staudinger and, in general, bythe German school led to a rapid industrialdevelopment in Germany. As early as 1931, IG
Farben started to market crystal polystyrene (GPPS,General Purpose PolyStyrene). The decisive thrust todevelop styrene materials, however, came with theSecond World War, when the difficulty of importingnatural rubber, due to naval blockades, drove theleading powers to increasing the production ofstyrene for use in synthetic rubbers. In the immediatepost-war period, the reopening of trade withcountries that produced natural lattices brought abouta marked excess capacity of styrene, which gave anirreversible boost to the growth of styrene-basedpolymeric materials. Thanks also to the economiceffects of the reconstruction, the commercial use ofanother key material, EPS (Expandable PolyStyrene),spread rapidly. In 1954 Dow started the industrialproduction of HIPS (High Impact PolyStyrene; Amoset al., 1954; Amos, 1974), which overcame thetraditional limitations of GPPS on mechanicalresistance. 1959 saw the first commercialization ofacrylonitrile-butadiene-styrene (ABS) ter-polymers.In the same period production of styrene-acrylonitrile (SAN) copolymers started. Table 1shows the composition of the main thermoplasticstyrenic polymers.
837VOLUME II / REFINING AND PETROCHEMICALS
12.4
Thermoplastic styrenic polymers
Polymer Styrene PB AN Expanding agent
GPPS (General Purpose PolyStyrene) yes – – –
SAN (Styrene-AcryloNitrile) yes – yes –
EPS (Expandable PolyStyrene) yes – – yes
HIPS (High Impact PolyStyrene) yes yes – –
ABS (Acrylonitrile-Butadiene-Styrene) yes yes yes –
Table 1. Definition and composition of the main thermoplastic styrenic polymers
PB, polybutadienic rubber; AN, acrylonitrile comonomer.

12.4.2 Chemistry of polymerization
The synthesis mechanisms for the most widespreadthermoplastic styrenic materials are shown below.
Polystyrene homopolymerGPPS is commonly produced worldwide by radical
reaction in the liquid phase. With this mechanismstyrene monomer polymerizes, in the absence ofoxygen or other inhibitors, even through simpleheating, since formation of radicals starting polymericchains occurs through an intermediate Diels-Alderreaction (a Diels-Alder adduct, i.e. 1-phenyl-1,2,3,8-tetrahydronaphthalene), which is unstable andobtained through the combination of two styrenemolecules (Mayo, 1968). Fig. 1 shows the complexreaction mechanism for the formation of radicals tostart the polymerization and the cyclic oligomers thatform alongside the styrene soluble polymer (and anyaromatic solvents present, in any percentage).
The products of the reaction scheme in Fig. 1,under industrial conditions, are mainly the cyclictrimers (four isomers, tPhcyH), in quantities that areabout 5-10 times less than the diphenylcyclobutanes(two isomers, dPhcyB). The radicals, which form invery low percentages, however, start the
polymerization reaction which produces chainscomposed of thousands of styrenic units, thus theoverall result obtained by heating styrene to 90-180°Cis polymer formation of over 99%.
In industrial production the rate of polymerizationcan be controlled by regulating the temperature andadding a radical initiator, usually a peroxide. In this wayit is possible to set the rate at which the chains begin,the propagation of which speeds up as the temperatureincreases. The termination of the free radical chainsmainly occurs by coupling and by transfer reaction tosmall molecules, which are the aforementionedintermediate dimer (Olaj, 1971) of the Diels-Alderreaction (DA-add) and possibly aromatic solvents orsubstances that are specifically added to control themolecular weight, such as, for example, mercaptans.The slow stage in the reaction is that of the formation ofthe first radicals, while propagation is rapid. To make achain some hundredths or a few tenths of a second aresufficient depending on the temperature.
In the simplified kinetic model of the styrene’spolymerization reaction shown below, which includesthe stages of the thermal and peroxide initiation,propagation, transfer to the monomer, solvent and anychain regulator, and termination by coupling anddisproportionation, the complex reaction of thermalinitiation of Fig. 1 is summarised in a single reactionand the transfer to the DA-adduct is traceable to themonomer transfer on the assumption that itsconcentration is proportional to that of styrene:
Initition I �� 2 R0�3 M �� 2 R0�R0� � M �� R1�
Propagation Rn� � M �� R�n�1Transfer Rn� � T, M, S �� Pn + T�, M�, S�Termination Rn� � Rm� �� Pn + Pm
Rn� � Rm� �� Pn�m
where I is the peroxide, M the styrene, R� the radical,T the chain transfer agent, S the solvent and P thepolymer.
The polymer chains obtained have a linear, atacticstructure. To synthesise chains with a branchedstructure it is necessary to adopt particular measures inthe formulation, such as the addition of divinyliccomonomers or polyfunctional initiators.
At temperatures over 100°C, as primarily used inindustrial manufacturing, the chain transfer to theintermediate DA-add avoids the appearance of thephenomenon of self-acceleration called the ‘gel’ or‘Trommsdorf effect’, which in the polymerization ofstyrene at low temperatures or for some styrenecopolymers results in an uncontrolled increase inreaction rate and molecular weight. For this reason thepolymerization of styrene is not critical, and run-away
838 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
�
�
�
�
�
. .
tPhcyH
dPhcyB
M
DA-add DA-R
SR
Tc
Fig. 1. Reaction mechanism for the formation ofpolymerization initiating radicals and cyclic oligomers. M, styrene; tPhcyH, four isomers; dPhcyB, two isomers;DA-add, Diels-Alder adduct; DA-R, radical of the Diels-Alderadduct; SR, 1-phenyl-ethyl-radical; Tc, cyclic trimers.
or unstable reactions do not occur in normal operatingconditions.
At temperatures over 200°C reactions involving thedepolymerization or degradation of the PS chainsformed or of the growing radical chains also becomeimportant (Campbell et al., 2003). Degradation of thePS leads to formation of styrene, diphenylbutene,triphenylhexene, a-methylstyrene and toluene. Styreneis produced by a mechanism that is inverse to that ofthe propagation, while the other compounds originatefrom a migration of the radical chain terminal withsubsequent rearrangement.
Styrene copolymers The free radical copolymerization of styrene with
vinylic monomers, such as AcryloNitrile (AN) andother derivates of acrylic and metacrylic acid, is widelyused for mass production of thermoplastic styrenicpolymers. Comonomers mixed with styrene polymerisein accordance with the well-known laws of radicalcopolymerization, which produces atactic polymerswith random monomer sequences (Ito and Yamashita,1965). The composition of a copolymer depends on thecomposition of the mix of monomers and the relativereactivity of the radical chains, which grow with themonomers present. In the scheme below are set out thekinetic equations of the propagation reactions for twomonomers in which the reactivity depends only on thelast monomer linked in the chain, which supports theradical (terminal model) and not on the composition ofthe rest of the chain (in this case two parameters knownas reactivity ratios are sufficient to determine theaverage composition of the polymer chains which formfrom a monomeric mix at a given composition):
PA� � A �� PAA� vaa�kaa[PA�][A]PA� � S �� PAS� vas�kas [PA�][S]PS� � S �� PSS� vss�kss [PS�][S]PS� � A �� PSA� vsa�ksa [PS�][A]
where v is the reaction rate and k the rate constant. Thereactivity ratios are defined as ra�vaa/vas and rs�vss/vsa.
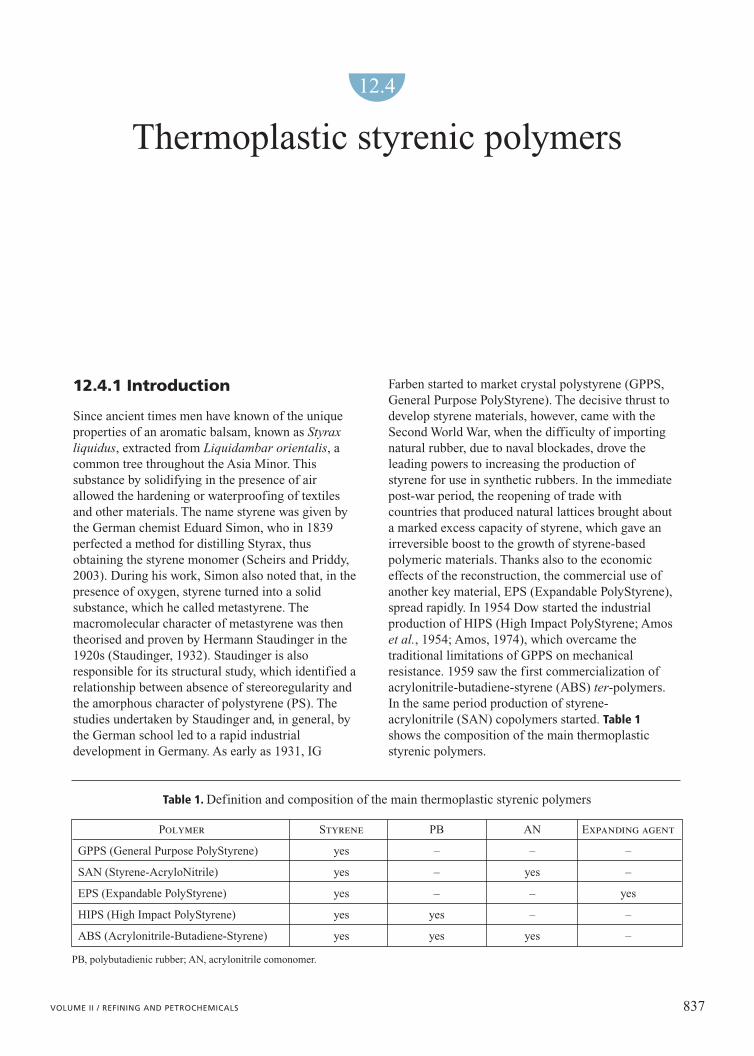
In the case that the reactivity of the radical chainsdepends also on the penultimate monomer linked tothe chain, the number of propagation reactions toconsider becomes eight and the reactivity ratios whichdetermine the composition of the copolymer are four.The graph in Fig. 2 depicts on the y-axis the compositionof the Styrene-AcryloNitrile (SAN) copolymer,calculated with the penultimate model (Ferrando andLongo, 1997), which is obtained in relation to themonomeric composition shown on the x-axis.
There is only one point in which the composition ofthe copolymer and that of the monomeric mix are thesame; as happens for binary equilibria of liquid andvapour, this point is called the azeotrope (23.8% in
weight of AN). Through probability tests it is possibleto extract formulae which, with the aforementionedreactivity ratios, determine the sequence of chainmonomers.
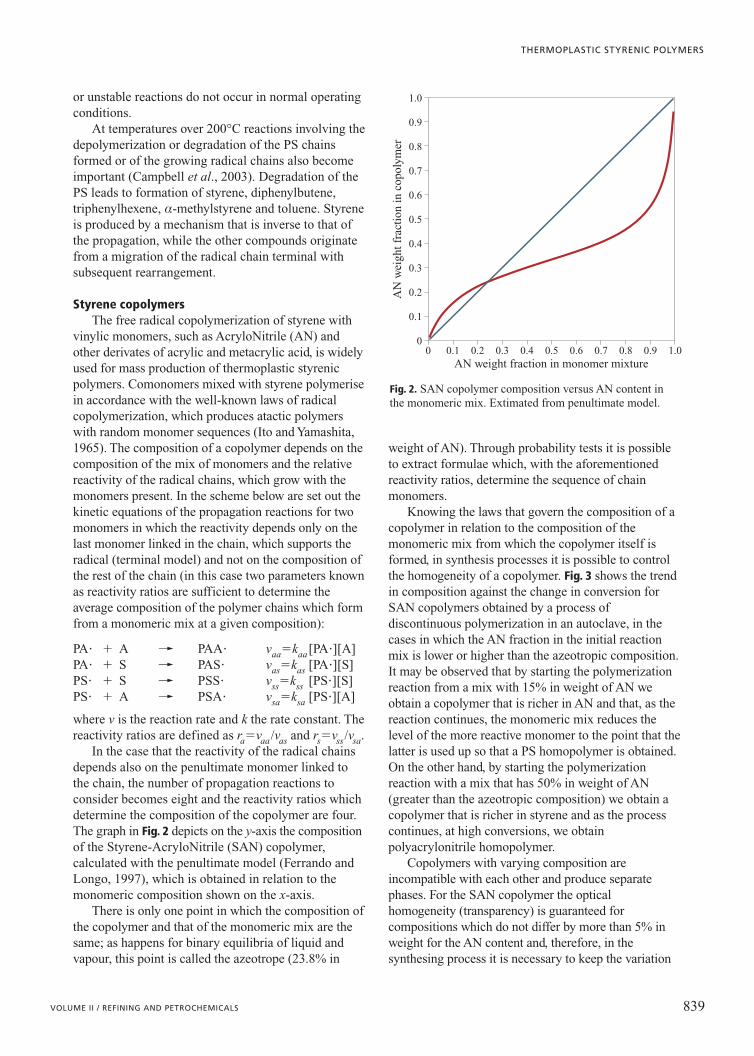
Knowing the laws that govern the composition of acopolymer in relation to the composition of themonomeric mix from which the copolymer itself isformed, in synthesis processes it is possible to controlthe homogeneity of a copolymer. Fig. 3 shows the trendin composition against the change in conversion forSAN copolymers obtained by a process ofdiscontinuous polymerization in an autoclave, in thecases in which the AN fraction in the initial reactionmix is lower or higher than the azeotropic composition.It may be observed that by starting the polymerizationreaction from a mix with 15% in weight of AN weobtain a copolymer that is richer in AN and that, as thereaction continues, the monomeric mix reduces thelevel of the more reactive monomer to the point that thelatter is used up so that a PS homopolymer is obtained.On the other hand, by starting the polymerizationreaction with a mix that has 50% in weight of AN(greater than the azeotropic composition) we obtain acopolymer that is richer in styrene and as the processcontinues, at high conversions, we obtainpolyacrylonitrile homopolymer.
Copolymers with varying composition areincompatible with each other and produce separatephases. For the SAN copolymer the opticalhomogeneity (transparency) is guaranteed forcompositions which do not differ by more than 5% inweight for the AN content and, therefore, in thesynthesing process it is necessary to keep the variation
839VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
AN
wei
ght f
ract
ion
in c
opol
ymer
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
AN weight fraction in monomer mixture0 0.1 0.2 0.4 0.6 0.80.3 0.5 0.7 0.9 1.0
Fig. 2. SAN copolymer composition versus AN content inthe monomeric mix. Extimated from penultimate model.
of the monomeric composition under control, in orderto guarantee the homogeneity of the copolymer.
Polyphasic styrenic polymersThe PS homopolymer and the acrylic copolymers
of styrene are widely used and appreciated for theirmechanical, thermal and processing properties. Inorder to reduce the fragility of styrenic polymers,which behave like amorphous, homogenous resins,with a glass transition temperature (Tg) which is higherthan the application temperature, an elastomer isintroduced with a Tg lower than the ambienttemperature (from �50 to �110°C), which has thepurpose of absorbing the impact energy byinterrupting the propagation of the fracture. Theelastomeric phase is incompatible with styrenic resinand generally consists of PolyButadiene (PBu) whichmust be emulsified in particles of 0.2-6 mm with goodadhesion to the interface. The mechanical, rheologicand thermal properties of styrenic materials dependlargely not only on the macroscopic chemicalcomposition, but also on the size and structure of thedispersed phase and the distribution of the molecularweights in the continuous phase. If the structure isvery fine and ordered, it is possible to obtaintransparent polyphasic polymers, since the light is notrefracted. Transparent shockproof materials are alsoobtained by acting on the composition of the phases sothat they have the same refractive index.
Mass production of HIPS and ABSThe mass production process for HIPS or ABS
involves the polymerization of styrene (or of styreneand acrylonitrile) in the presence of polybutadiene,polystyrene and PS-PBu block copolymer in ahomogeneous solution in batch reactors.
When between 4 and 6% of styrene is polymerizedthere is a phase separation, in which the continuousphase consists of the PBu solution in styrene and thedispersed phase of a PS solution in styrene. At amicroscopic level there is an emulsion of PS styrenicsolution in the PBu styrenic solution, where theemulsifier is the PS-PBu block copolymer. As thereaction progresses, PS is produced until the volumeof the PS solution in styrene exceeds the volume of thePBu solution in styrene and phase inversion occurs,accompanied by an evident change in the viscosity ofthe solution.
From this point on, the dispersed phase consists ofPBu with free occluded PS homopolymer and PSgrafted on PBu. As the polymerization reactionprogresses there is also a partial reticulation of thePBu which tends to freeze the structure and the size ofthe particles in the elastomeric dispersed phase. Thereticulation reaction of the PBu mainly occurs in thefinishing phase, in which, at temperatures over 200°C,the polymer is separated from the residual monomerand solvent. The following table sets out the reactionswhich involve the PBu in the polymerization reactionof the styrene and which may be added to the tableabove for homopolymerization:
R0� + PBu �� PBu� + RH
Rn� + PBu �� PBu� + Pn
PBu� + M �� PBuR1�
PBuRn� + M �� PBuR�n�1
PBu� + PBu� �� PBurt
PBuRn� + Rm� �� PBuPn�m
PBuRn� + Rm� �� PBuPn + Pm
PBuRn� + PBuRm� �� PBuPn�mPBu
PBuRn� + PBuRm� �� PBuPn + PBuPm
840 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60 70 80 90 100
AN
con
tent
(w
eigh
t % )
monomers conversion (%)
monomer
monomer
instantaneouspolymer
instantaneous polymer
cumulativepolymer
cumulative polymer
Fig. 3. Composition of the instantaneous and cumulative SANcopolymer versusmonomers conversion ina discontinuous process(batch) for mixes with15% (solid line) and50% (dashed line)weight of AN in themonomer feed.
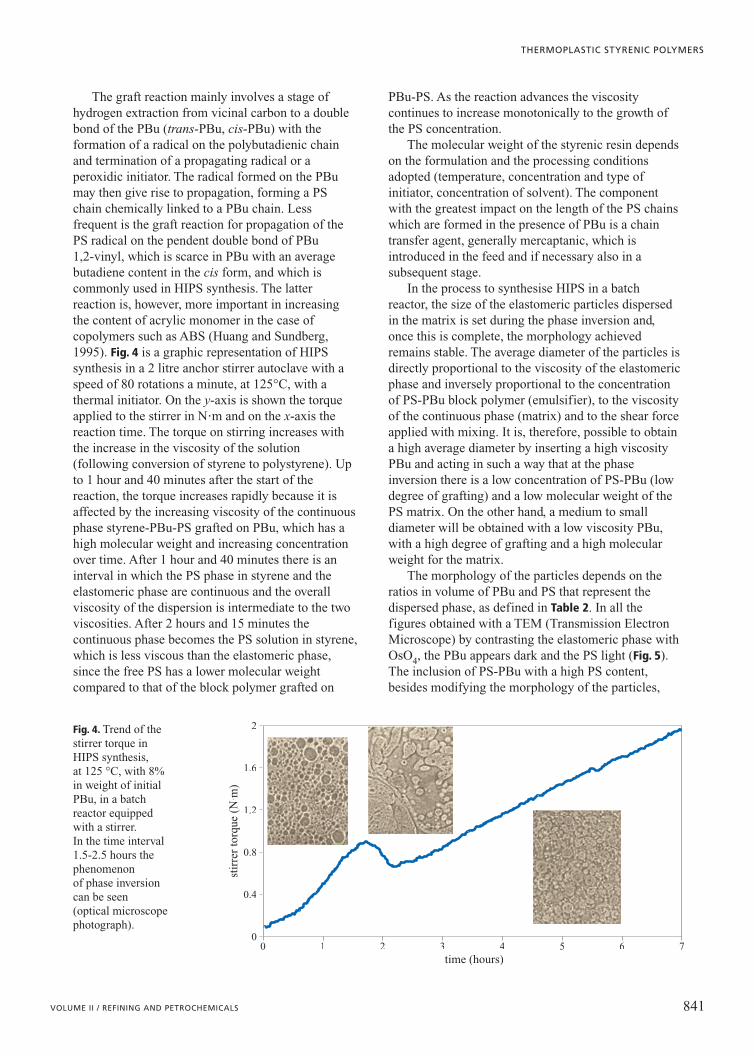
The graft reaction mainly involves a stage ofhydrogen extraction from vicinal carbon to a doublebond of the PBu (trans-PBu, cis-PBu) with theformation of a radical on the polybutadienic chainand termination of a propagating radical or aperoxidic initiator. The radical formed on the PBumay then give rise to propagation, forming a PSchain chemically linked to a PBu chain. Lessfrequent is the graft reaction for propagation of thePS radical on the pendent double bond of PBu 1,2-vinyl, which is scarce in PBu with an averagebutadiene content in the cis form, and which iscommonly used in HIPS synthesis. The latterreaction is, however, more important in increasingthe content of acrylic monomer in the case ofcopolymers such as ABS (Huang and Sundberg,1995). Fig. 4 is a graphic representation of HIPSsynthesis in a 2 litre anchor stirrer autoclave with aspeed of 80 rotations a minute, at 125°C, with athermal initiator. On the y-axis is shown the torqueapplied to the stirrer in N�m and on the x-axis thereaction time. The torque on stirring increases withthe increase in the viscosity of the solution(following conversion of styrene to polystyrene). Upto 1 hour and 40 minutes after the start of thereaction, the torque increases rapidly because it isaffected by the increasing viscosity of the continuousphase styrene-PBu-PS grafted on PBu, which has ahigh molecular weight and increasing concentrationover time. After 1 hour and 40 minutes there is aninterval in which the PS phase in styrene and theelastomeric phase are continuous and the overallviscosity of the dispersion is intermediate to the twoviscosities. After 2 hours and 15 minutes thecontinuous phase becomes the PS solution in styrene,which is less viscous than the elastomeric phase,since the free PS has a lower molecular weightcompared to that of the block polymer grafted on
PBu-PS. As the reaction advances the viscositycontinues to increase monotonically to the growth ofthe PS concentration.
The molecular weight of the styrenic resin dependson the formulation and the processing conditionsadopted (temperature, concentration and type ofinitiator, concentration of solvent). The componentwith the greatest impact on the length of the PS chainswhich are formed in the presence of PBu is a chaintransfer agent, generally mercaptanic, which isintroduced in the feed and if necessary also in asubsequent stage.
In the process to synthesise HIPS in a batchreactor, the size of the elastomeric particles dispersedin the matrix is set during the phase inversion and,once this is complete, the morphology achievedremains stable. The average diameter of the particles isdirectly proportional to the viscosity of the elastomericphase and inversely proportional to the concentrationof PS-PBu block polymer (emulsifier), to the viscosityof the continuous phase (matrix) and to the shear forceapplied with mixing. It is, therefore, possible to obtaina high average diameter by inserting a high viscosityPBu and acting in such a way that at the phaseinversion there is a low concentration of PS-PBu (lowdegree of grafting) and a low molecular weight of thePS matrix. On the other hand, a medium to smalldiameter will be obtained with a low viscosity PBu,with a high degree of grafting and a high molecularweight for the matrix.
The morphology of the particles depends on theratios in volume of PBu and PS that represent thedispersed phase, as defined in Table 2. In all thefigures obtained with a TEM (Transmission ElectronMicroscope) by contrasting the elastomeric phase withOsO4, the PBu appears dark and the PS light (Fig. 5).The inclusion of PS-PBu with a high PS content,besides modifying the morphology of the particles,
841VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
stir
rer
torq
ue (
N. m
)
0
0.4
0.8
1.2
1.6
2
time (hours)0 1 32 54 6 7
Fig. 4. Trend of thestirrer torque inHIPS synthesis, at 125 °C, with 8%in weight of initialPBu, in a batchreactor equippedwith a stirrer. In the time interval1.5-2.5 hours thephenomenon of phase inversioncan be seen (optical microscopephotograph).
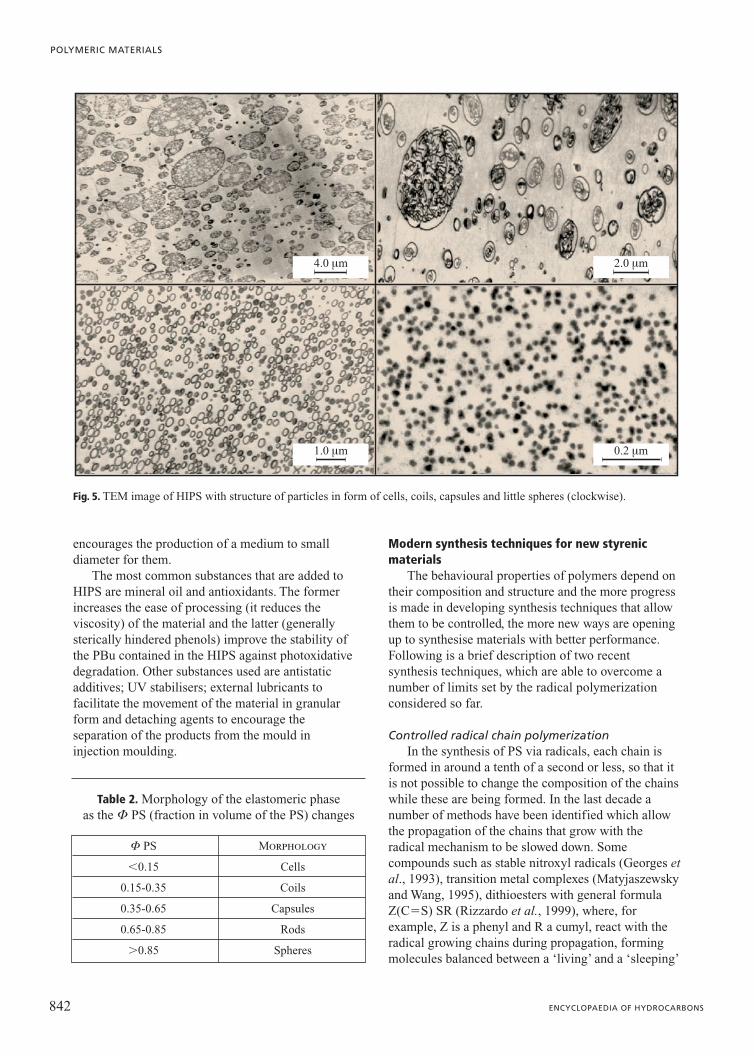
encourages the production of a medium to smalldiameter for them.
The most common substances that are added toHIPS are mineral oil and antioxidants. The formerincreases the ease of processing (it reduces theviscosity) of the material and the latter (generallysterically hindered phenols) improve the stability ofthe PBu contained in the HIPS against photoxidativedegradation. Other substances used are antistaticadditives; UV stabilisers; external lubricants tofacilitate the movement of the material in granularform and detaching agents to encourage theseparation of the products from the mould ininjection moulding.
Modern synthesis techniques for new styrenicmaterials
The behavioural properties of polymers depend ontheir composition and structure and the more progressis made in developing synthesis techniques that allowthem to be controlled, the more new ways are openingup to synthesise materials with better performance.Following is a brief description of two recentsynthesis techniques, which are able to overcome anumber of limits set by the radical polymerizationconsidered so far.
Controlled radical chain polymerizationIn the synthesis of PS via radicals, each chain is
formed in around a tenth of a second or less, so that itis not possible to change the composition of the chainswhile these are being formed. In the last decade anumber of methods have been identified which allowthe propagation of the chains that grow with theradical mechanism to be slowed down. Somecompounds such as stable nitroxyl radicals (Georges etal., 1993), transition metal complexes (Matyjaszewskyand Wang, 1995), dithioesters with general formulaZ(C�S) SR (Rizzardo et al., 1999), where, forexample, Z is a phenyl and R a cumyl, react with theradical growing chains during propagation, formingmolecules balanced between a ‘living’ and a ‘sleeping’
842 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
4.0 mm 2.0 mm
1.0 mm 0.2 mm
Fig. 5. TEM image of HIPS with structure of particles in form of cells, coils, capsules and little spheres (clockwise).
F PS Morphology
�0.15 Cells
0.15-0.35 Coils
0.35-0.65 Capsules
0.65-0.85 Rods
�0.85 Spheres
Table 2. Morphology of the elastomeric phaseas the F PS (fraction in volume of the PS) changes
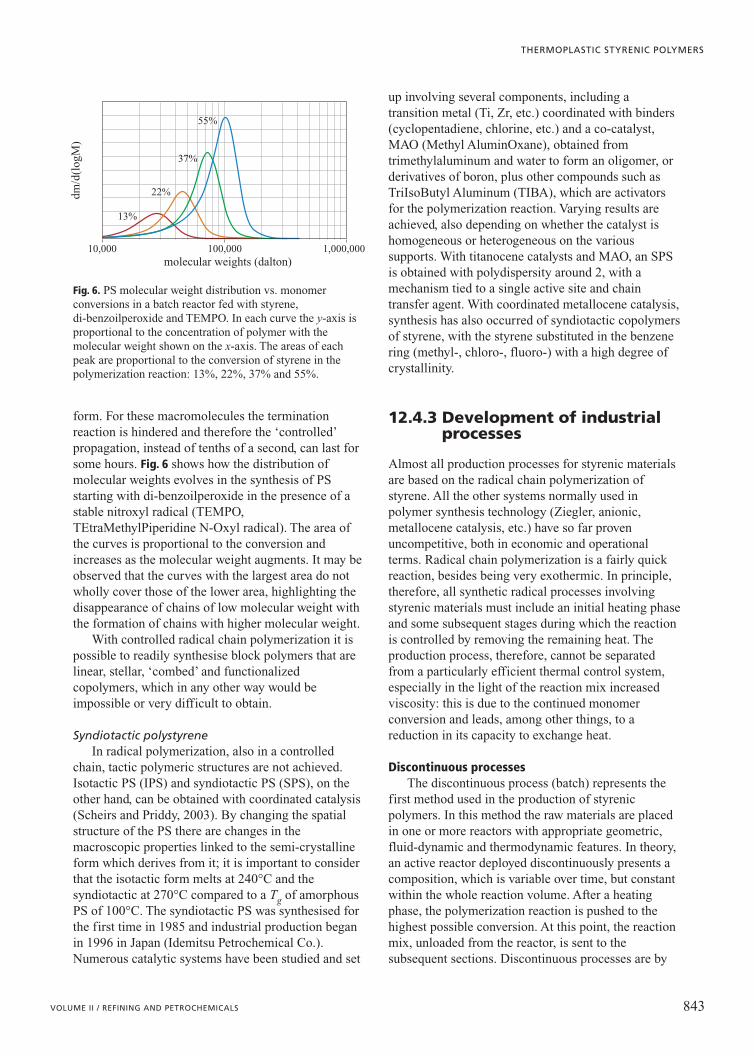
form. For these macromolecules the terminationreaction is hindered and therefore the ‘controlled’propagation, instead of tenths of a second, can last forsome hours. Fig. 6 shows how the distribution ofmolecular weights evolves in the synthesis of PSstarting with di-benzoilperoxide in the presence of astable nitroxyl radical (TEMPO,TEtraMethylPiperidine N-Oxyl radical). The area ofthe curves is proportional to the conversion andincreases as the molecular weight augments. It may beobserved that the curves with the largest area do notwholly cover those of the lower area, highlighting thedisappearance of chains of low molecular weight withthe formation of chains with higher molecular weight.
With controlled radical chain polymerization it ispossible to readily synthesise block polymers that arelinear, stellar, ‘combed’ and functionalizedcopolymers, which in any other way would beimpossible or very difficult to obtain.
Syndiotactic polystyreneIn radical polymerization, also in a controlled
chain, tactic polymeric structures are not achieved.Isotactic PS (IPS) and syndiotactic PS (SPS), on theother hand, can be obtained with coordinated catalysis(Scheirs and Priddy, 2003). By changing the spatialstructure of the PS there are changes in themacroscopic properties linked to the semi-crystallineform which derives from it; it is important to considerthat the isotactic form melts at 240°C and thesyndiotactic at 270°C compared to a Tg of amorphousPS of 100°C. The syndiotactic PS was synthesised forthe first time in 1985 and industrial production beganin 1996 in Japan (Idemitsu Petrochemical Co.).Numerous catalytic systems have been studied and set
up involving several components, including atransition metal (Ti, Zr, etc.) coordinated with binders(cyclopentadiene, chlorine, etc.) and a co-catalyst,MAO (Methyl AluminOxane), obtained fromtrimethylaluminum and water to form an oligomer, orderivatives of boron, plus other compounds such asTriIsoButyl Aluminum (TIBA), which are activatorsfor the polymerization reaction. Varying results areachieved, also depending on whether the catalyst ishomogeneous or heterogeneous on the varioussupports. With titanocene catalysts and MAO, an SPSis obtained with polydispersity around 2, with amechanism tied to a single active site and chaintransfer agent. With coordinated metallocene catalysis,synthesis has also occurred of syndiotactic copolymersof styrene, with the styrene substituted in the benzenering (methyl-, chloro-, fluoro-) with a high degree ofcrystallinity.
12.4.3 Development of industrialprocesses
Almost all production processes for styrenic materialsare based on the radical chain polymerization ofstyrene. All the other systems normally used inpolymer synthesis technology (Ziegler, anionic,metallocene catalysis, etc.) have so far provenuncompetitive, both in economic and operationalterms. Radical chain polymerization is a fairly quickreaction, besides being very exothermic. In principle,therefore, all synthetic radical processes involvingstyrenic materials must include an initial heating phaseand some subsequent stages during which the reactionis controlled by removing the remaining heat. Theproduction process, therefore, cannot be separatedfrom a particularly efficient thermal control system,especially in the light of the reaction mix increasedviscosity: this is due to the continued monomerconversion and leads, among other things, to areduction in its capacity to exchange heat.
Discontinuous processesThe discontinuous process (batch) represents the
first method used in the production of styrenicpolymers. In this method the raw materials are placedin one or more reactors with appropriate geometric,fluid-dynamic and thermodynamic features. In theory,an active reactor deployed discontinuously presents acomposition, which is variable over time, but constantwithin the whole reaction volume. After a heatingphase, the polymerization reaction is pushed to thehighest possible conversion. At this point, the reactionmix, unloaded from the reactor, is sent to thesubsequent sections. Discontinuous processes are by
843VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
55%
37%
22%
13%
dm/d
(log
M)
molecular weights (dalton)10,000 100,000 1,000,000
Fig. 6. PS molecular weight distribution vs. monomerconversions in a batch reactor fed with styrene, di-benzoilperoxide and TEMPO. In each curve the y-axis isproportional to the concentration of polymer with themolecular weight shown on the x-axis. The areas of eachpeak are proportional to the conversion of styrene in thepolymerization reaction: 13%, 22%, 37% and 55%.

their very nature particularly flexible and adaptable tovarying production needs, even if they perform poorlyin terms of productivity and operating costs.
Discontinuous mass polymerizationThe pioneering phase of polymerization processes
was based essentially on a simple but barely efficientsystem. The technology, known as can process, in factinvolved heating the styrene in metal cans; after a fewdays, the polymer formed at almost total conversionwas extracted from the cans and ground. However, itwas soon evident that it was necessary to introduce apre-polymerization phase (Fig. 7) in a discontinuousreactor, run at a low temperature (80°C) but for a verylong time (48 hours). In this way it was possible tomanage the most critical phase of the reaction, i.e. thatin which the monomer has higher concentrations, inmild conditions. When conversion reached around35%, the mix was then transferred into an adiabaticreactor composed of various sectors (‘trays’). Once itwas completely converted, the product was extractedand ground.
A further improvement was made through theintroduction of a semi-continuous technology, whichallowed more rational control of the reaction, as wellas a considerable reduction in production times. Themix arising from the pre-polymerisers was transferredto a tubular reactor (‘tower’), in which the temperaturewas brought from 100°C at the entrance up to 180°Con exit, thus allowing the reaction to proceed up to aconversion rate of around 99%. The tubular reactorwas continuously discharged from the bottom, and thepolymer directly pellettized without purificationoperations (devolatilization).
The mass processes described previously, besidesthe typical problems of discontinuous technologies inrelation to time and costs, brought very seriousproblems of homogeneity and stirring of the reactivemass. A decisive improvement in this sense came withthe introduction of the process in suspension (Fig. 8),in which the control of the heat developed by the
reaction is guaranteed by a fluid, with features such asnot to interfere with the polymerization (generallywater), present within a batch reactor.
Polymerization, therefore, occurs within monomerparticles suspended in water, thanks also to the use ofsuitable (tensioactive) suspending agents, which canbe added at various stages of conversion so as tomodify the size distribution of the beads. It is possiblein this phase to use initiators or chain transfer agents.When the conversion is almost complete, the product,in the form of beads, is separated by centrifuge fromthe water solution and then air-dried.
Suspension technology saw certain popularity inthe 1950s, by virtue of its aforementioned features(good control of the viscosity and of the thermaldevelopment of the system). In the next decade,paramount needs in terms of environment, economicsand quality would lead (see below) to the almostcomplete substitution of the suspension process withthat of continuous mass production in the synthesis ofcompact styrenic polymers. This technology is stillprevalent where the move to continuous technologyhas still not occurred, in other words in the field ofExpandable PolyStyrene (EPS). In this case, duringpolymerization an organic expanding agent (typicallypentane) is incorporated, leading, in the subsequentphases of processing, to the expansion of the bead and,therefore, to the cellular structure, with a subsequentreduction in the density of the product.
Emulsion polymerizationIn parallel to the development of the suspension
process, a further technology is taking hold whichfinds its ideal application only in the synthesis ofbiphasic materials, in which, in addition to theaforementioned problems of the polymerization ofstyrene, there are difficulties linked to the graftingreaction between the macromolecular chain and therubber phase.
Synthesis via emulsion (Fig. 9) is a discontinuousprocess characterised by various stages.
844 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
styreneinitiator
water/vapour
water/vapour
additives
pre-polymerizator tray reactor mill storage
inert
Fig. 7. Process of discontinuous mass production withtray reactor.
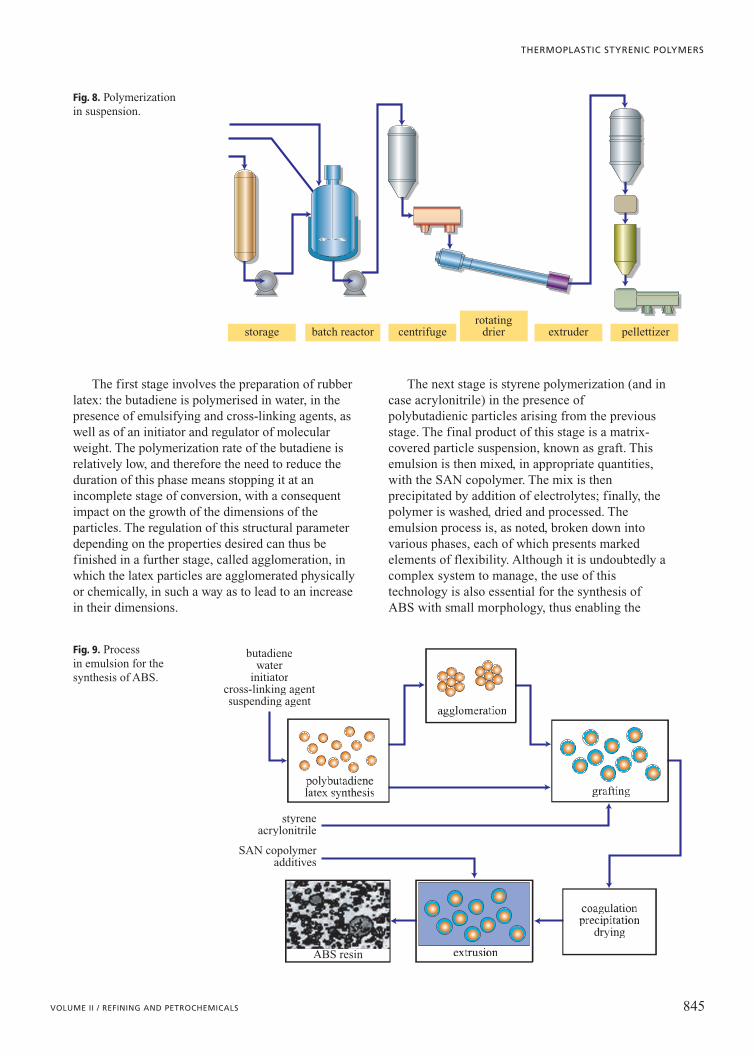
The first stage involves the preparation of rubberlatex: the butadiene is polymerised in water, in thepresence of emulsifying and cross-linking agents, aswell as of an initiator and regulator of molecularweight. The polymerization rate of the butadiene isrelatively low, and therefore the need to reduce theduration of this phase means stopping it at anincomplete stage of conversion, with a consequentimpact on the growth of the dimensions of theparticles. The regulation of this structural parameterdepending on the properties desired can thus befinished in a further stage, called agglomeration, inwhich the latex particles are agglomerated physicallyor chemically, in such a way as to lead to an increasein their dimensions.
The next stage is styrene polymerization (and incase acrylonitrile) in the presence ofpolybutadienic particles arising from the previousstage. The final product of this stage is a matrix-covered particle suspension, known as graft. Thisemulsion is then mixed, in appropriate quantities,with the SAN copolymer. The mix is thenprecipitated by addition of electrolytes; finally, thepolymer is washed, dried and processed. Theemulsion process is, as noted, broken down intovarious phases, each of which presents markedelements of flexibility. Although it is undoubtedly acomplex system to manage, the use of thistechnology is also essential for the synthesis ofABS with small morphology, thus enabling the
845VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
pellettizerextruderrotating
driercentrifugebatch reactorstorage
butadienewater
initiatorcross-linking agentsuspending agent
styreneacrylonitrile
SAN copolymeradditives
ABS resin
Fig. 8. Polymerization in suspension.
Fig. 9. Process in emulsion for thesynthesis of ABS.

achievement of rubber phase particles with anaverage diameter of less than 0.2 microns.
For this reason the emulsion process has not beensignificantly used in the HIPS field, where the balanceof structure to properties requires a marked increase inthe size of the particles. Another argument against theuse of the emulsion system in the production of HIPSis the difficulty of controlling the rubber phase cross-linking, a particularly important parameter for themechanical properties of high-impact polystyrene.
Continuous processesContrarily to the processes considered so far, a
continuous stationary process is based on thecontinuous feedstock of the raw materials and on thecontinuous output of products, together with sub-products or processing residues. Generally, thisprocedure permits to optimise key parameters such asinvestment costs, quality constancy of the productsand production capacity. Unlike for other polymers,above all for polyethylene, for which over time thewidest range of continuous processes have beendeveloped (high pressure, solution, slurry, gas-phase,etc.), the physical state of the raw materials used forthe synthesis of styrenic polymers has led from thevery start to mass production technology.
Continuous mass processAs highlighted in the previous paragraphs,
discontinuous technologies, while flexible and usefulfor the synthesis of particular materials (for exampleglossy ABS), present undoubted problems from theoperational and financial viewpoint. In recent decadesenvironmental issues have been becoming clearer andincreasingly unavoidable in relation to the quality ofthe finished product (monomers and residualsubstances) and production features (quantity andquality of effluents, emissions in the workplace, etc.).
The continuous mass production process does notuse raw materials or additives outside the reaction(such as water in the aforementioned processes), andthus allows a drastic reduction in the quantity ofemissions. This technology is thus normally conceivedas a closed cycle; everything that has not reacted at theend of the polymerization section is separated out inthe devolatilization section, condensed and feed onceagain at the entrance of the system. Only a minimalamount of purge, which in the most efficient processescan be around 1.0-1.5%, is made necessary to removenon-polymerisable matters that otherwise would tendto accumulate.
The traditional model for continuous mass process(Fig. 10) involves a rubber dissolution section (for twophase polymers), where the polybutadiene is dissolvedin styrene, a reaction section, which generally involves
various reaction stages in a series, and adevolatilization section (in one or more stages), fromwhich the purified polymer is sent to a pellettiser.Polymerization takes place in the presence of a solvent(generally ethyl benzene) that limits the viscosity ofthe system in case of uncontrolled reactions, as well ascontaining the reaction velocity.
Two different kind of reactors are generally used incontinuous mass process: CSTR (Continuous StirredTank Reactor) and PFR (Plug Flow Reactor).
CSTR is a fully mixed reactor and therefore thereaction velocity, the concentration and thetemperature are the same in every point of the reactor.This means that the output conditions are exactly thesame as those in the reactor itself. In the synthesis ofstyrenic polymers the removal of heat is provided bythe evaporation of part of the reaction mix. Among themain advantages of CSTR may be included the relativeease of cleaning, the good thermal control and the lowconstruction costs, while the main disadvantage is thelow specific productivity.
The PFR may be compared to a tube throughwhich the reaction mix passes without any mixing inan axial sense (no backward mixing). Along thedirection of the flow, every section has uniformfeatures in terms of speed, composition andtemperature. This system is particularly suitable forprocesses where there is rubber and, therefore, whereit is necessary to have effective control of the phaseinversion.
Compared to the CSTR, the PFR is more efficientin terms of specific productivity, even if the need toguarantee the control of the reaction by means of heatexchange elements makes the investment costgenerally higher.
Both types of reactors used in the continuous massprocessing envisage the output from thepolymerization section of a mix (pre-polymer)composed of polymer (65-75%), unreacted monomerand non-polymerisable matters (solvent), at atemperature between 140 and 170°C.
The pre-polymer is then further heated in a pre-heater, where a diathermic oil system provides theheat needed both to reach a temperature that isnormally above 200°C (sensible heat) and to keep thetemperature of the polymer constant during theevaporation of the non-polymeric components (latentheat of vaporization). Given the relatively lowvolatility of the involved compounds, in order to limitthe thermal stress on the product the devolatilizationprocess takes place under reduced pressure.
In addition, the design of the pre-heater is ofparticular importance as it must allow the distributionof the polymers over an exchange surface as wide aspossible, albeit using small equipment and without
846 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS

causing excessive pressure drops. The process ofdevolatilization can consist in one or two stages atvarying pressures. In this second case, most of thevolatile fractions are separated from the polymer in thehighest pressure stage (1.6-2.0 kPa), thus facilitatingthe process of condensation which precedes therecycling at the top of the reaction train. In this phasethe concentration of residual styrene is 2,000-3,000ppm. In the second stage, which operates under moresevere vacuum conditions (0.3-0.4 kPa), thepurification process is completed, leading to residualmonomer content values generally below 500 ppm.
12.4.4 Structure and properties
Structure of polystyrene homopolymer The macromolecule of polystyrene, in the form
that is commonly obtained by radical polymerization,has an atactic configuration. In the solid state (i.e.below the glass transition temperature) PS is thereforean amorphous and transparent glassy polymer.
The glass transition temperature (Tg), measured byDSC (Differential Scanning Calorimetry) isapproximately 100°C (Brandrup and Immergut, 1989).As for all thermoplastic polymers, the value of Tg,independent of the degree of polymerization when thelatter is high, decreases if the chain length fallsbeneath a critical value. For PS this occurs formolecular weight values smaller than about 100,000(degree of polymerization approximately equal to1,000; Claudy et al., 1983).
The density of PS in the glassy state and at roomtemperature is approximately 1.05 g/cm3. Thedependence of density on temperature (thermal
expansion volume coefficient) is 2.7�10�4 cm3/g/Kbelow Tg and 6.0�10�4 cm3/g/K above Tg (Brandrupand Immergut, 1989; van Krevelen, 1990).
The polymeric chain, because of the large benzenering, is relatively inflexible. A direct consequence ofthis is that the molecular weight between theentanglements, Me (the term entanglements identifiesthe points of intersection or crossover between themacromolecular chains) has a relatively high value(approximately 19,000) if compared with those of theother most common thermoplastics (Ferry, 1980;Donald and Kramer, 1982b). This structural feature isimportant in determining the properties of thematerial, as will be described in the following.
For many commercial grades of PS, a commonpractice is the addition of plasticisers, usually paraffinoils, in amounts not exceeding 5 or 6 weight %, themain purpose of which is to reduce the viscosity of thepolymer melt at processing temperatures. Thestructural effect of a plasticiser in a polymer is anincrease of the free volume (Ferry, 1980), which leadsto a reduction in the Tg. The paraffin oils used in PSdetermine a lowering of Tg of approximately 4°C forevery percentage unit with additives by weight.
Rheological properties
ViscosityThe first important rheological consequence of the
molecular structure concerns the dependence of theNewtonian viscosity h0 (the limit value of viscosity asthe shear flow rate tends to zero) of the polymer melton the molecular weight M. As for all macromolecularfluids (Ferry, 1980), below a critical molecular weightvalue Mc the viscosity increases linearly with
847VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
vacuumsystem
finishedproduct
condensation
furnacecombustionfumes
fuel
styrene � rubber(dissolution)
additives
styrenechemicals
ethylbenzeneperoxideadditives
additives
feedstock treatment pre-polymerization polymerization devolatilization pelletizing
Fig. 10. Process in continuous mass production for the synthesis of HIPS.
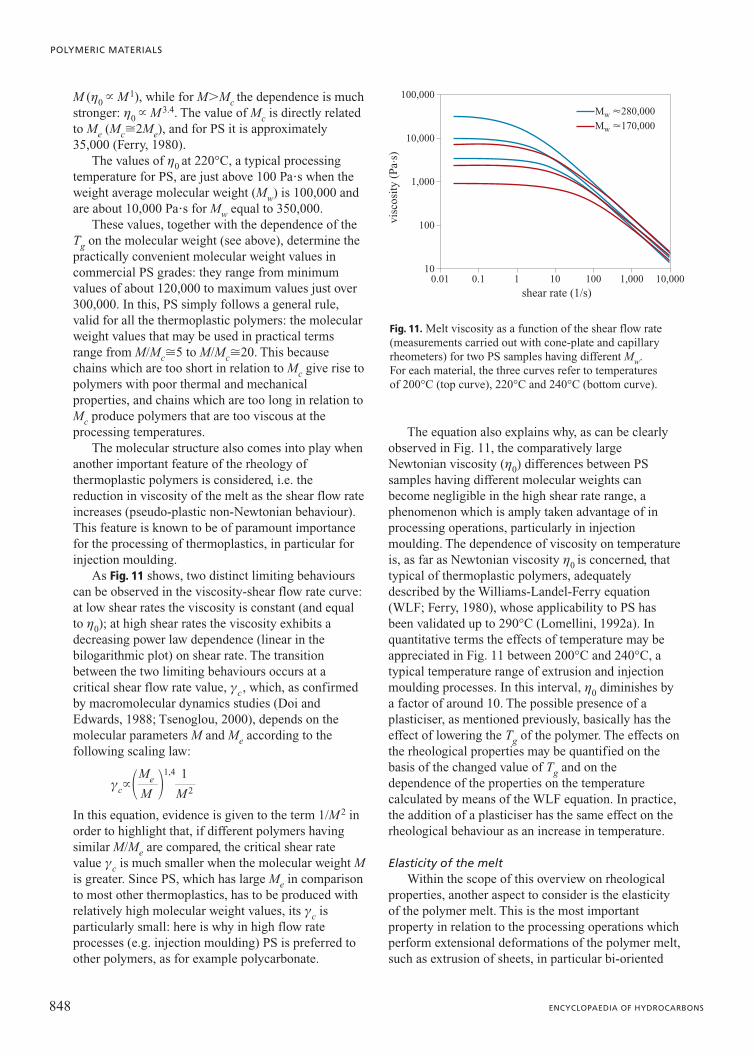
M (h0 � M1), while for M�Mc the dependence is muchstronger: h0 � M3.4. The value of Mc is directly relatedto Me (Mc�2Me), and for PS it is approximately35,000 (Ferry, 1980).
The values of h0 at 220°C, a typical processingtemperature for PS, are just above 100 Pa�s when theweight average molecular weight (Mw) is 100,000 andare about 10,000 Pa�s for Mw equal to 350,000.
These values, together with the dependence of theTg on the molecular weight (see above), determine thepractically convenient molecular weight values incommercial PS grades: they range from minimumvalues of about 120,000 to maximum values just over300,000. In this, PS simply follows a general rule,valid for all the thermoplastic polymers: the molecularweight values that may be used in practical termsrange from M/Mc�5 to M/Mc�20. This becausechains which are too short in relation to Mc give rise topolymers with poor thermal and mechanicalproperties, and chains which are too long in relation toMc produce polymers that are too viscous at theprocessing temperatures.
The molecular structure also comes into play whenanother important feature of the rheology ofthermoplastic polymers is considered, i.e. thereduction in viscosity of the melt as the shear flow rateincreases (pseudo-plastic non-Newtonian behaviour).This feature is known to be of paramount importancefor the processing of thermoplastics, in particular forinjection moulding.
As Fig. 11 shows, two distinct limiting behaviourscan be observed in the viscosity-shear flow rate curve:at low shear rates the viscosity is constant (and equalto h0); at high shear rates the viscosity exhibits adecreasing power law dependence (linear in thebilogarithmic plot) on shear rate. The transitionbetween the two limiting behaviours occurs at acritical shear flow rate value, gc, which, as confirmedby macromolecular dynamics studies (Doi andEdwards, 1988; Tsenoglou, 2000), depends on themolecular parameters M and Me according to thefollowing scaling law:
Me 1gc��12�
1,412
M M2
In this equation, evidence is given to the term 1/M2 inorder to highlight that, if different polymers havingsimilar M/Me are compared, the critical shear ratevalue gc is much smaller when the molecular weight Mis greater. Since PS, which has large Me in comparisonto most other thermoplastics, has to be produced withrelatively high molecular weight values, its gc isparticularly small: here is why in high flow rateprocesses (e.g. injection moulding) PS is preferred toother polymers, as for example polycarbonate.
The equation also explains why, as can be clearlyobserved in Fig. 11, the comparatively largeNewtonian viscosity (h0) differences between PSsamples having different molecular weights canbecome negligible in the high shear rate range, aphenomenon which is amply taken advantage of inprocessing operations, particularly in injectionmoulding. The dependence of viscosity on temperatureis, as far as Newtonian viscosity h0 is concerned, thattypical of thermoplastic polymers, adequatelydescribed by the Williams-Landel-Ferry equation(WLF; Ferry, 1980), whose applicability to PS hasbeen validated up to 290°C (Lomellini, 1992a). Inquantitative terms the effects of temperature may beappreciated in Fig. 11 between 200°C and 240°C, atypical temperature range of extrusion and injectionmoulding processes. In this interval, h0 diminishes bya factor of around 10. The possible presence of aplasticiser, as mentioned previously, basically has theeffect of lowering the Tg of the polymer. The effects onthe rheological properties may be quantified on thebasis of the changed value of Tg and on thedependence of the properties on the temperaturecalculated by means of the WLF equation. In practice,the addition of a plasticiser has the same effect on therheological behaviour as an increase in temperature.
Elasticity of the meltWithin the scope of this overview on rheological
properties, another aspect to consider is the elasticityof the polymer melt. This is the most importantproperty in relation to the processing operations whichperform extensional deformations of the polymer melt,such as extrusion of sheets, in particular bi-oriented
848 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
visc
osit
y (P
a.s)
10
100
1,000
10,000
100,000
shear rate (1/s)
Mw �280,000
Mw �170,000
0.01 0.1 1 10 100 1,000 10,000
Fig. 11. Melt viscosity as a function of the shear flow rate(measurements carried out with cone-plate and capillaryrheometers) for two PS samples having different Mw. For each material, the three curves refer to temperatures of 200°C (top curve), 220°C and 240°C (bottom curve).
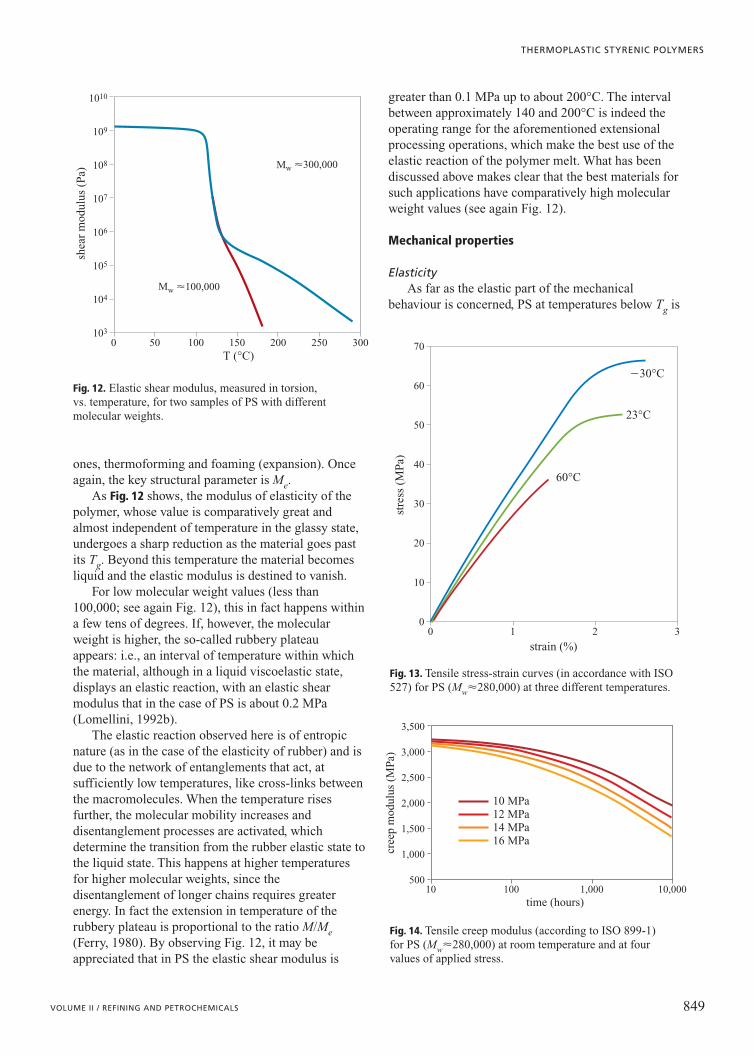
ones, thermoforming and foaming (expansion). Onceagain, the key structural parameter is Me.
As Fig. 12 shows, the modulus of elasticity of thepolymer, whose value is comparatively great andalmost independent of temperature in the glassy state,undergoes a sharp reduction as the material goes pastits Tg. Beyond this temperature the material becomesliquid and the elastic modulus is destined to vanish.
For low molecular weight values (less than100,000; see again Fig. 12), this in fact happens withina few tens of degrees. If, however, the molecularweight is higher, the so-called rubbery plateauappears: i.e., an interval of temperature within whichthe material, although in a liquid viscoelastic state,displays an elastic reaction, with an elastic shearmodulus that in the case of PS is about 0.2 MPa(Lomellini, 1992b).
The elastic reaction observed here is of entropicnature (as in the case of the elasticity of rubber) and isdue to the network of entanglements that act, atsufficiently low temperatures, like cross-links betweenthe macromolecules. When the temperature risesfurther, the molecular mobility increases anddisentanglement processes are activated, whichdetermine the transition from the rubber elastic state tothe liquid state. This happens at higher temperaturesfor higher molecular weights, since thedisentanglement of longer chains requires greaterenergy. In fact the extension in temperature of therubbery plateau is proportional to the ratio M/Me(Ferry, 1980). By observing Fig. 12, it may beappreciated that in PS the elastic shear modulus is
greater than 0.1 MPa up to about 200°C. The intervalbetween approximately 140 and 200°C is indeed theoperating range for the aforementioned extensionalprocessing operations, which make the best use of theelastic reaction of the polymer melt. What has beendiscussed above makes clear that the best materials forsuch applications have comparatively high molecularweight values (see again Fig. 12).
Mechanical properties
ElasticityAs far as the elastic part of the mechanical
behaviour is concerned, PS at temperatures below Tg is
849VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
shea
r m
odul
us (
Pa)
103
104
105
106
107
108
109
1010
T (°C)0 50 100 150 200 300250
Mw �300,000
Mw �100,000
Fig. 12. Elastic shear modulus, measured in torsion, vs. temperature, for two samples of PS with differentmolecular weights.
0
10
20
30
40
50
60
70
0 1 2 3
stre
ss (
MPa
)
strain (%)
�30°C
23°C
60°C
Fig. 13. Tensile stress-strain curves (in accordance with ISO527) for PS (Mw�280,000) at three different temperatures.
cree
p m
odul
us (
MPa
)
500
1,000
1,500
2,000
2,500
3,000
3,500
time (hours)
10 MPa12 MPa14 MPa16 MPa
10 100 1,000 10,000
Fig. 14. Tensile creep modulus (according to ISO 899-1) for PS (Mw�280,000) at room temperature and at fourvalues of applied stress.

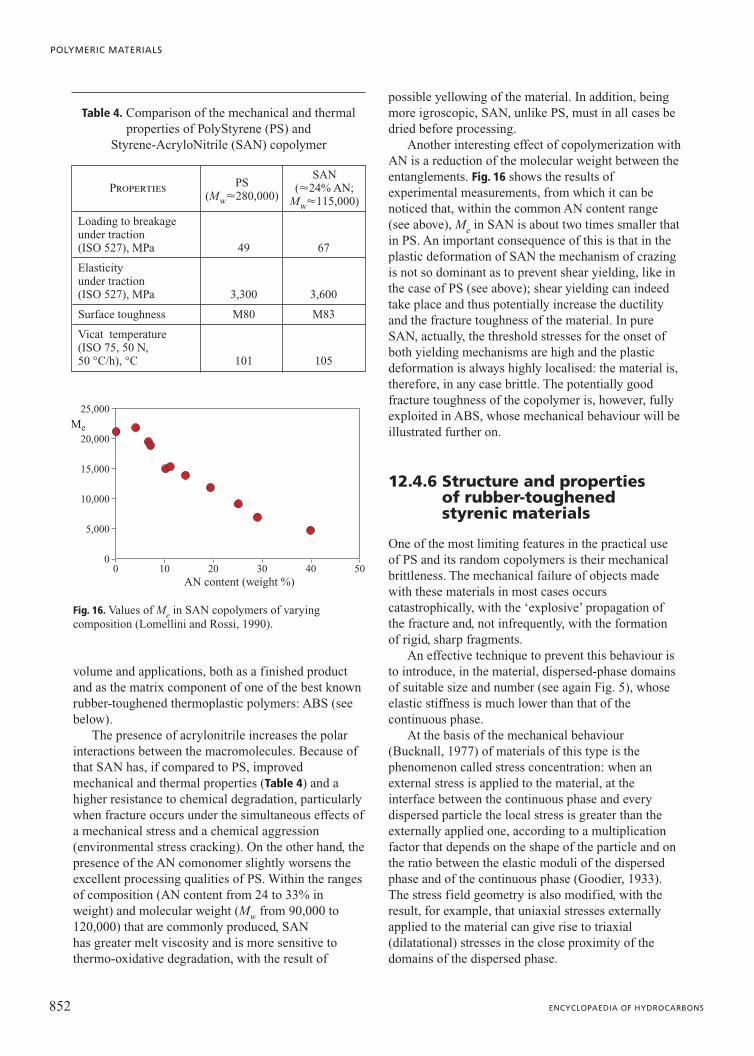
a typical amorphous polymer in the glassy state and assuch has, at room temperature, a tensile modulus ofelasticity of approximately 3,300 MPa and a Poissoncoefficient of about 0.35 (Brandrup and Immergut,1989; van Krevelen, 1990). The elastic properties forT�Tg are largely independent of the molecular weight.
Of course PS, like all polymeric materials, exhibitsa markedly viscoelastic behaviour even in the glassstate. A consequence of this is a strong dependence ofall the mechanical properties, and in particular of theelastic ones, on temperature and time.
Figs. 13 and 14 illustrate respectively the temperaturedependence of the tensile stress-strain curve and thetime evolution of the creep modulus (the term creeprefers to the deformation, increasing with time, of amaterial subjected to a constant load). In Fig. 14 thenon-linearity of the elastic behaviour of the material canalso be observed: for a fixed time value, the creepmodulus diminishes as the applied force increases.
Plastic deformation and fractureUnlike elastic properties, values related to the
resistance to plastic deformation and fracture arestrongly affected by the molecular weight. This haslong been known as an experimental evidence: a studydated to 1959 (McCormick et al., 1959) reports thevalues of tensile stress and strain at break and thevalues of absorbed energy in tensile-impact tests forsamples of PS covering a broad range of molecularweights. The results of this work show that mechanicalresistance improves strongly with increasing molecularweight for Mw values included between about 40,000and 120,000, and then flattens down, tending to leveloff for Mw greater than 200,000. These figures werelargely confirmed by a subsequent work (Hauss,1969).
In order to explain these experimentalobservations, it is necessary to analyse the details ofthe plastic deformation processes in the material. It iswell known that, in glassy amorphous thermoplasticpolymers, plastic deformation can take place throughtwo basic mechanisms (Haward and Young, 1997):• Shear yielding, where deformation occurs at a
constant volume, with the appearing of typicalshear bands and macroscopic necking. In this case,the behaviour is usually ductile, with relativelylarge deformations before fracture.
• Crazing, in which case the appearing of typicaldefects, called crazes, is observed. Crazes extendon planes perpendicular to the applied stress andare similar to cracks, but unlike these they containa thick network of microscopic fibrils, whichbridge their two surfaces (Kramer, 1983). Crazingis commonly associated with a brittle mechanicalbehaviour.
PS behaves in a distinct manner for what concernsplastic deformation: driven by the tensile componentof the applied stress, the mechanism which comes intoplay is in all cases crazing, for which the onset stress‘threshold’ is, in the case of this particular polymer,always lower (under traction) than that for the shearyielding mechanism.
There is a precise correlation between themolecular structure of PS and its definite propensity todeform by crazing. It has been shown (Donald andKramer, 1982b; Kramer, 1983) that the most importantfeature in this case is the maximum extensibility (lmax)of the amorphous macromolecular structure. The lattercan be depicted as a three-dimensional ‘network’,whose nodes are the points of entanglement and whosemesh threads are the chain segments between twosuccessive entanglements. With reference to themolecular weight of the monomeric unit M0, theextensibility lmax is given by the ratio between thelength of the mesh thread, which is proportional toMe/M0, and the distance between the nodes ‘at rest’,which instead is proportional to (Me/M0)
1/2. Hence, inthe comparison between different polymers, lmaxvaries in proportion to (Me/M0)
1/2. The above citedauthors show the experimental correlation betweenlmax (and therefore Me/M0) and the prevalence of thecrazing mechanism over that of shear yielding: inpolymers having a large lmax crazing is dominant, andvice versa. As previously underlined, PS has one of thegreatest values of Me, and therefore also of Me/M0 andof lmax when compared to other commonly usedthermoplastic polymers. This is the reason of the cleardominance of the crazing mechanism in this material.
In a number of papers, Kramer and co-workersexplained further important details of PS crazing interms of structure-properties relationships.
Crazing, as mentioned earlier, is associated withbrittleness. Under strain, the craze extends and itswalls move apart through the progressive transfer ofbulk material into the fibrils, but this allows only smallmacroscopic deformations before the onset of fibrilsbreakdown causes the catastrophic transformation ofthe craze into a crack.
By means of TEM analysis of PS thin filmsstrained in tension, it has been possible to measure(Kramer and Berger, 1990), on samples with varyingMw, critical strain values for the nucleation (ec) and thefracture (ef) of the crazes. While ec is independent ofthe molecular weight, ef is strongly affected by thechain length and increases from values comparable toec when Mw is about 40,000 (the freshly nucleatedcraze breaks) to values as large as 10ec when Mw is200,000, remaining then constant if Mw is furtherincreased. These results can be explained on the basisof the need of a fully developed entanglement network
850 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS

for the fibrillar structure to reach its maximumstability. Molecular weights up to around 2Me give riseto minimal fracture resistance values; the fracturetoughness then rises sharply with the increasing chainlength, to reach a maximum when the molecularweight is around 10Me. These results confirm andexplain the empirical relations between Mw and themechanical resistance of PS mentioned above(McCormick et al., 1959; Hauss, 1969).
12.4.5 Structure and properties of random copolymers of styrene
A general feature of random copolymers is that thevalues of their properties are intermediate betweenthose of the homopolymers based on the constituentmonomers. Through random copolymerization, it is,therefore, sometimes possible to obtain interesting‘compromises’, in which it is possible to provide forspecific deficiencies of a homopolymer without losingtoo much of its positive characteristics. In the case ofstyrene, important copolymers, since they areproduced industrially and are present in the appliedfield, are the styrene maleic anhydride copolymer(SMA), the styrene-methyl methacrylate copolymer(SMMA), and the styrene-acrylonitrile (SAN)copolymer.
Styrene-maleic anhydrideThe possibility of copolymerising styrene and
maleic anhydride (MA) has been long known (Scheirsand Priddy, 2003). The most interesting property of the
copolymer is the glass transition temperature, whichincreases markedly with the increasing MA content.
Fig. 15 shows a plot of the Vicat softeningtemperature (measured in accordance with ISO 306)as a function of the content in weight of MA. The costof such a marked gain in thermal resistance is,however, a significant increase in the melt viscosity: interms of a standard melt fluidity test, the Melt FlowRate, measured at 220°C with a load of 10 kg inaccordance with ISO 1133, the values can change,from about 48.5 g/10 min when the content of MA is6.7 wt% to around 9.4 g/10 min when the MA is 18.9wt%. Moreover, as further consequences of thepresence of maleic anhydride, the copolymer haspoorer mechanical strength and a more pronouncedyellowing with respect to homopolymer PS.
Styrene-methyl methacrylatePolyMethylMethAcrylate (PMMA) is appreciated
for its excellent optical properties, for its surfacehardness (which, for example, gives rise to its goodresistance to scratching) and for its resistance to ultra-violet radiation. On the other hand, PMMA isnotoriously susceptible to chemical degradation byalcohols, which limits its use in food-contactapplications.
SMMA copolymers have superior optical andsurface properties with respect to PS and resistalcohols better than PMMA. Table 3 shows the valuesof some relevant properties for a typical SMMA(containing around 70% methylmethacrylate) and forthe two homopolymers in comparison. The possibilityto adjust the refractive index of the material by varyingthe comonomer content, make SMMA copolymersused in the synthesis of transparent rubber reinforcedmaterials.
Styrene-acrylonitrileSAN is, within the family of styrene-based random
copolymers, by far the most important for production
851VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
100
110
120
130
140
150
160
0 5 10 15 20 25 30
Vic
at s
ofte
ning
tem
pera
ture
(°C
)
maleic anhydride content (weight %)
Fig. 15. Vicat Softening Temperature (VST), measured in accordance with ISO 306 for styrene-maleic anhydridecopolymers as a function of the content of MA comonomer(Rossi, 1984).
Properties PMMA PS SMMA
Refraction index 1.491 1.590 1.564
Abbe number 57.2 30.8 35.0
Transmittance 92 87-92 90
Surface toughness M 97 M 70 M 75
Scratch resistance 0.3 2.2 1.7
Table 3. Comparison between the properties in PolyMethylMethAcrylate (PMMA),
PolyStyrene (PS) and SMMA copolymer containingaround 70% of MMA
volume and applications, both as a finished productand as the matrix component of one of the best knownrubber-toughened thermoplastic polymers: ABS (seebelow).
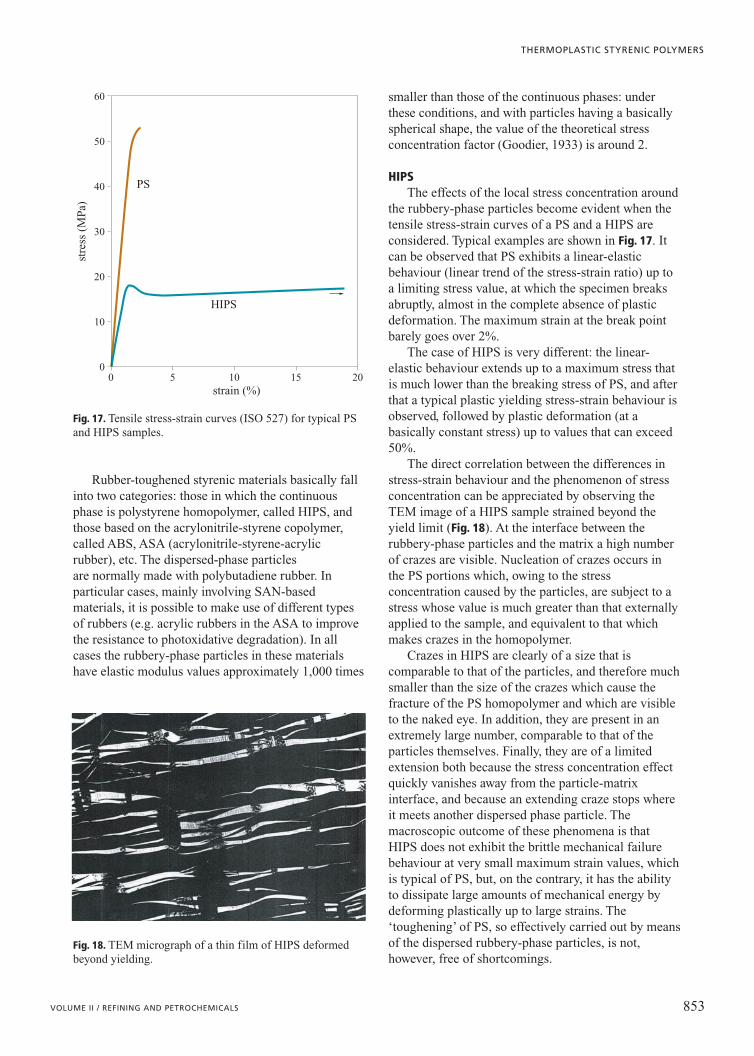
The presence of acrylonitrile increases the polarinteractions between the macromolecules. Because ofthat SAN has, if compared to PS, improvedmechanical and thermal properties (Table 4) and ahigher resistance to chemical degradation, particularlywhen fracture occurs under the simultaneous effects ofa mechanical stress and a chemical aggression(environmental stress cracking). On the other hand, thepresence of the AN comonomer slightly worsens theexcellent processing qualities of PS. Within the rangesof composition (AN content from 24 to 33% inweight) and molecular weight (Mw from 90,000 to120,000) that are commonly produced, SAN has greater melt viscosity and is more sensitive tothermo-oxidative degradation, with the result of
possible yellowing of the material. In addition, beingmore igroscopic, SAN, unlike PS, must in all cases bedried before processing.
Another interesting effect of copolymerization withAN is a reduction of the molecular weight between theentanglements. Fig. 16 shows the results ofexperimental measurements, from which it can benoticed that, within the common AN content range(see above), Me in SAN is about two times smaller thatin PS. An important consequence of this is that in theplastic deformation of SAN the mechanism of crazingis not so dominant as to prevent shear yielding, like inthe case of PS (see above); shear yielding can indeedtake place and thus potentially increase the ductilityand the fracture toughness of the material. In pureSAN, actually, the threshold stresses for the onset ofboth yielding mechanisms are high and the plasticdeformation is always highly localised: the material is,therefore, in any case brittle. The potentially goodfracture toughness of the copolymer is, however, fullyexploited in ABS, whose mechanical behaviour will beillustrated further on.
12.4.6 Structure and properties of rubber-toughenedstyrenic materials
One of the most limiting features in the practical useof PS and its random copolymers is their mechanicalbrittleness. The mechanical failure of objects madewith these materials in most cases occurscatastrophically, with the ‘explosive’ propagation ofthe fracture and, not infrequently, with the formationof rigid, sharp fragments.
An effective technique to prevent this behaviour isto introduce, in the material, dispersed-phase domainsof suitable size and number (see again Fig. 5), whoseelastic stiffness is much lower than that of thecontinuous phase.
At the basis of the mechanical behaviour(Bucknall, 1977) of materials of this type is thephenomenon called stress concentration: when anexternal stress is applied to the material, at theinterface between the continuous phase and everydispersed particle the local stress is greater than theexternally applied one, according to a multiplicationfactor that depends on the shape of the particle and onthe ratio between the elastic moduli of the dispersedphase and of the continuous phase (Goodier, 1933).The stress field geometry is also modified, with theresult, for example, that uniaxial stresses externallyapplied to the material can give rise to triaxial(dilatational) stresses in the close proximity of thedomains of the dispersed phase.
852 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
Me
0
25,000
20,000
15,000
10,000
5,000
AN content (weight %)0 10 20 30 40 50
Fig. 16. Values of Me in SAN copolymers of varyingcomposition (Lomellini and Rossi, 1990).
PSSAN
Properties(Mw�280,000)
(�24% AN;Mw�115,000)
Loading to breakage under traction (ISO 527), MPa 49 67
Elasticity under traction(ISO 527), MPa 3,300 3,600
Surface toughness M80 M83
Vicat temperature (ISO 75, 50 N,50 °C/h), °C 101 105
Table 4. Comparison of the mechanical and thermalproperties of PolyStyrene (PS) and
Styrene-AcryloNitrile (SAN) copolymer
Rubber-toughened styrenic materials basically fallinto two categories: those in which the continuousphase is polystyrene homopolymer, called HIPS, andthose based on the acrylonitrile-styrene copolymer,called ABS, ASA (acrylonitrile-styrene-acrylicrubber), etc. The dispersed-phase particles are normally made with polybutadiene rubber. Inparticular cases, mainly involving SAN-basedmaterials, it is possible to make use of different typesof rubbers (e.g. acrylic rubbers in the ASA to improvethe resistance to photoxidative degradation). In allcases the rubbery-phase particles in these materialshave elastic modulus values approximately 1,000 times
smaller than those of the continuous phases: underthese conditions, and with particles having a basicallyspherical shape, the value of the theoretical stressconcentration factor (Goodier, 1933) is around 2.
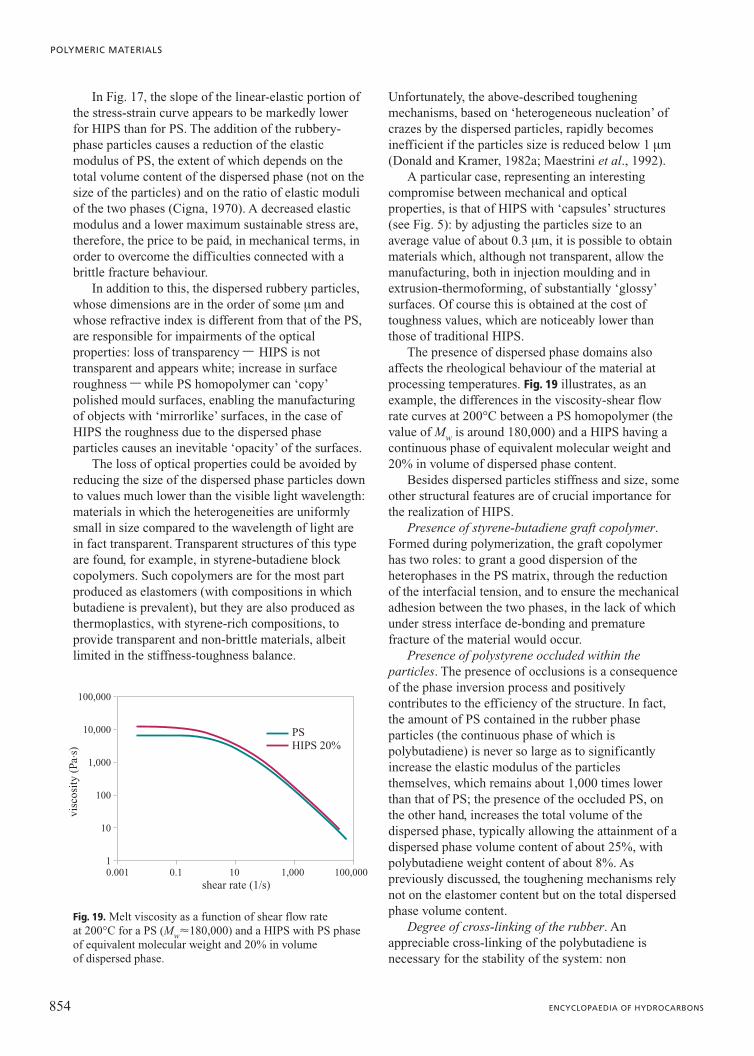
HIPSThe effects of the local stress concentration around
the rubbery-phase particles become evident when thetensile stress-strain curves of a PS and a HIPS areconsidered. Typical examples are shown in Fig. 17. Itcan be observed that PS exhibits a linear-elasticbehaviour (linear trend of the stress-strain ratio) up toa limiting stress value, at which the specimen breaksabruptly, almost in the complete absence of plasticdeformation. The maximum strain at the break pointbarely goes over 2%.
The case of HIPS is very different: the linear-elastic behaviour extends up to a maximum stress thatis much lower than the breaking stress of PS, and afterthat a typical plastic yielding stress-strain behaviour isobserved, followed by plastic deformation (at abasically constant stress) up to values that can exceed50%.
The direct correlation between the differences instress-strain behaviour and the phenomenon of stressconcentration can be appreciated by observing theTEM image of a HIPS sample strained beyond theyield limit (Fig. 18). At the interface between therubbery-phase particles and the matrix a high numberof crazes are visible. Nucleation of crazes occurs inthe PS portions which, owing to the stressconcentration caused by the particles, are subject to astress whose value is much greater than that externallyapplied to the sample, and equivalent to that whichmakes crazes in the homopolymer.
Crazes in HIPS are clearly of a size that iscomparable to that of the particles, and therefore muchsmaller than the size of the crazes which cause thefracture of the PS homopolymer and which are visibleto the naked eye. In addition, they are present in anextremely large number, comparable to that of theparticles themselves. Finally, they are of a limitedextension both because the stress concentration effectquickly vanishes away from the particle-matrixinterface, and because an extending craze stops whereit meets another dispersed phase particle. Themacroscopic outcome of these phenomena is thatHIPS does not exhibit the brittle mechanical failurebehaviour at very small maximum strain values, whichis typical of PS, but, on the contrary, it has the abilityto dissipate large amounts of mechanical energy bydeforming plastically up to large strains. The‘toughening’ of PS, so effectively carried out by meansof the dispersed rubbery-phase particles, is not,however, free of shortcomings.
853VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
Fig. 17. Tensile stress-strain curves (ISO 527) for typical PSand HIPS samples.
Fig. 18. TEM micrograph of a thin film of HIPS deformedbeyond yielding.
0
10
20
30
40
50
60
0 5
PS
HIPS
10 15 20
stre
ss (
MPa
)
strain (%)
In Fig. 17, the slope of the linear-elastic portion ofthe stress-strain curve appears to be markedly lowerfor HIPS than for PS. The addition of the rubbery-phase particles causes a reduction of the elasticmodulus of PS, the extent of which depends on thetotal volume content of the dispersed phase (not on thesize of the particles) and on the ratio of elastic moduliof the two phases (Cigna, 1970). A decreased elasticmodulus and a lower maximum sustainable stress are,therefore, the price to be paid, in mechanical terms, inorder to overcome the difficulties connected with abrittle fracture behaviour.
In addition to this, the dispersed rubbery particles,whose dimensions are in the order of some mm andwhose refractive index is different from that of the PS,are responsible for impairments of the opticalproperties: loss of transparency – HIPS is nottransparent and appears white; increase in surfaceroughness – while PS homopolymer can ‘copy’polished mould surfaces, enabling the manufacturingof objects with ‘mirrorlike’ surfaces, in the case ofHIPS the roughness due to the dispersed phaseparticles causes an inevitable ‘opacity’ of the surfaces.
The loss of optical properties could be avoided byreducing the size of the dispersed phase particles downto values much lower than the visible light wavelength:materials in which the heterogeneities are uniformlysmall in size compared to the wavelength of light arein fact transparent. Transparent structures of this typeare found, for example, in styrene-butadiene blockcopolymers. Such copolymers are for the most partproduced as elastomers (with compositions in whichbutadiene is prevalent), but they are also produced asthermoplastics, with styrene-rich compositions, toprovide transparent and non-brittle materials, albeitlimited in the stiffness-toughness balance.
Unfortunately, the above-described tougheningmechanisms, based on ‘heterogeneous nucleation’ ofcrazes by the dispersed particles, rapidly becomesinefficient if the particles size is reduced below 1 mm(Donald and Kramer, 1982a; Maestrini et al., 1992).
A particular case, representing an interestingcompromise between mechanical and opticalproperties, is that of HIPS with ‘capsules’ structures(see Fig. 5): by adjusting the particles size to anaverage value of about 0.3 mm, it is possible to obtainmaterials which, although not transparent, allow themanufacturing, both in injection moulding and inextrusion-thermoforming, of substantially ‘glossy’surfaces. Of course this is obtained at the cost oftoughness values, which are noticeably lower thanthose of traditional HIPS.
The presence of dispersed phase domains alsoaffects the rheological behaviour of the material atprocessing temperatures. Fig. 19 illustrates, as anexample, the differences in the viscosity-shear flowrate curves at 200°C between a PS homopolymer (thevalue of Mw is around 180,000) and a HIPS having acontinuous phase of equivalent molecular weight and20% in volume of dispersed phase content.
Besides dispersed particles stiffness and size, someother structural features are of crucial importance forthe realization of HIPS.
Presence of styrene-butadiene graft copolymer.Formed during polymerization, the graft copolymerhas two roles: to grant a good dispersion of theheterophases in the PS matrix, through the reductionof the interfacial tension, and to ensure the mechanicaladhesion between the two phases, in the lack of whichunder stress interface de-bonding and prematurefracture of the material would occur.
Presence of polystyrene occluded within theparticles. The presence of occlusions is a consequenceof the phase inversion process and positivelycontributes to the efficiency of the structure. In fact,the amount of PS contained in the rubber phaseparticles (the continuous phase of which ispolybutadiene) is never so large as to significantlyincrease the elastic modulus of the particlesthemselves, which remains about 1,000 times lowerthan that of PS; the presence of the occluded PS, onthe other hand, increases the total volume of thedispersed phase, typically allowing the attainment of adispersed phase volume content of about 25%, withpolybutadiene weight content of about 8%. Aspreviously discussed, the toughening mechanisms relynot on the elastomer content but on the total dispersedphase volume content.
Degree of cross-linking of the rubber. Anappreciable cross-linking of the polybutadiene isnecessary for the stability of the system: non
854 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
visc
osit
y (P
a.s)
1
10
100
1,000
10,000
100,000
shear rate (1/s)
PSHIPS 20%
0.001 0.1 10 1,000 100,000
Fig. 19. Melt viscosity as a function of shear flow rate at 200°C for a PS (Mw�180,000) and a HIPS with PS phase of equivalent molecular weight and 20% in volume of dispersed phase.

cross-linked particles would be destroyed duringprocessing. As far as fracture toughness is concerned,moreover, cross-linking of the rubber has an importantand complex role, since it affects the elastic propertiesof the rubber itself and its behaviour under the largestrain levels that it experiences during the PS-matrixmicro-crazing process above described. The finalproperties of the material are strongly affected bythese structural parameters, which have to beoptimised in view of the performances required by thevarious applications.
ABSThe structure of ABS is qualitatively similar to that
of HIPS, both when it is produced by the emulsionprocess (first used for production of this material), andwhen it is obtained by the continuous-mass process,which is increasingly widespread today. Similarly tothe case of HIPS, the basic structure is a glassy, brittlepolymeric matrix, which for ABS is a SAN copolymer,in which nearly spherical particles, mainly consistingof polybutadiene rubber, are dispersed. At the interfacebetween the phases, like in HIPS, there is a graftcopolymer, in this case SAN-polybutadiene.
The toughening mechanism is, therefore, also herebased on the stress concentration around the particles.While, however, in HIPS the ‘heterogeneousnucleation’ of plastic deformation can just occurthrough crazing, the only possible mechanism, in ABSshear yielding comes also into play, it being active inSAN for the reasons described previously. Theavailability of both plastic deformation mechanisms,which may be simultaneously activated, has twoimportant consequences: the rubbery-phase structuresand morphologies which maximise toughness in ABSare quantitatively different from the case of HIPS; thepotential of ABS, in terms of mechanical performance,is greater than that of HIPS.
As for the first point, it may be useful to brieflyreview some details about plastic deformation inrubber-toughened polymers, which have beenextensively discussed in the recent literature and arecurrently active research topics.
First of all, the nucleation of the shear yieldingmechanism by the rubbery-phase particles does notseem to be conditioned by a critical particles sizevalue, as in the case of crazing. The onset of shearplastic deformation appears to be triggered by theformation of cavities in the rubber particles (Bucknallet al., 1989; Ramsteiner and Heckmann, 1985): underthe dilatational component of the ‘concentrated’ stress(see above) existing around the particles, cavities cannucleate and grow in the rubber which, by modifyingthe stress field in the neighbouring continuous phase,make it favourable for shear yielding (Lazzeri andBucknall, 1995). This phenomenon can occur inparticles that are smaller than the critical size forcraze nucleation and it does not depend on theparticles size but on the appropriate values of othercharacteristics such as rubber cross-linking andrubber-matrix interfacial adhesion. For the efficientactivation of the shear yielding mechanism in ABS,optimum particles sizes are in the range from a fewtenths of mm up to 1 mm.
Crazing mechanism in the SAN matrix alsodisplays different features in comparison to the case ofPS. The critical size of the particles for crazesnucleation is smaller in SAN (Walker and Collyer,1994); in addition, the role of obstacles to crazepropagation, which in HIPS only the rubbery-phaseparticles can perform – and for this reason also theirsize must be sufficiently large (Maestrini et al., 1992),in ABS is mainly accomplished by shear yield zoneswhich grow at the crazes tip (Donald, 1994). Theoverall result of these micromechanical differences isthat optimum toughening in ABS is achieved withdispersed phase particles that are markedly smallerthan the typical particles of HIPS, in particular withsize distributions that include values in the order of 0.1mm, suitable for initiating shear yielding, and values ofabout 1 mm, appropriate for craze nucleation.
Figs. 20 A and B show two examples of dispersedphase morphologies in ABS produced by emulsion andmass processes respectively. They are only examples:in commercially-available products it is possible tofind large differences in the characteristics of the
855VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
1 mm 2 mm
a b
Fig. 20. TEMmicrographs of an ABSproduced by the emulsionpolymerization process(A) and an ABSproduced by thecontinuous-masspolymerization process (B).
A B
dispersed phases, which are a subject of constantresearch and improvement by the various producingcompanies.
The different mechanical performances of ABSand HIPS can be adequately described on the basis ofthe results of standard tensile tests (e.g. in accordancewith ISO 527) and impact strength tests (for examplein accordance with ISO 180): typically ABS,compared to HIPS, has greater elastic modulus andhigher yield stress and yield strain values; therefore, itcan be used for the manufacturing of objects which, onan equal size basis, are more rigid and can supportgreater loads. Plastic deformation following yielding,which in HIPS is homogeneously spread throughoutthe material, in ABS tends to localise (necking istypically observed during a tensile test), giving rise tosmaller nominal tensile strain at break values. Thisresult is only apparently negative: actually, it is a direct
manifestation of the active presence of the shear yieldmechanism which, by associating with crazing, givesrise in ABS to a greater ability to dissipate mechanicalenergy before fracture: the Izod impact strength,measured in accordance with ISO 180, ranges from 15kJ/m2 up to over 30 kJ/m2 for ABS, while it does notexceed 10 kJ/m2 for common HIPS grades.
The smaller size of the dispersed particles in ABSalso leads to lower surface roughness in comparison toHIPS and, therefore, to the possibility of mouldingobjects with highly glossy surfaces. On the other handsmaller particles are, at a given total rubbery-phasevolume content, closer to each other, with a greatereffect (in comparison to HIPS) of the dispersed phaseon the rheological properties of the material.
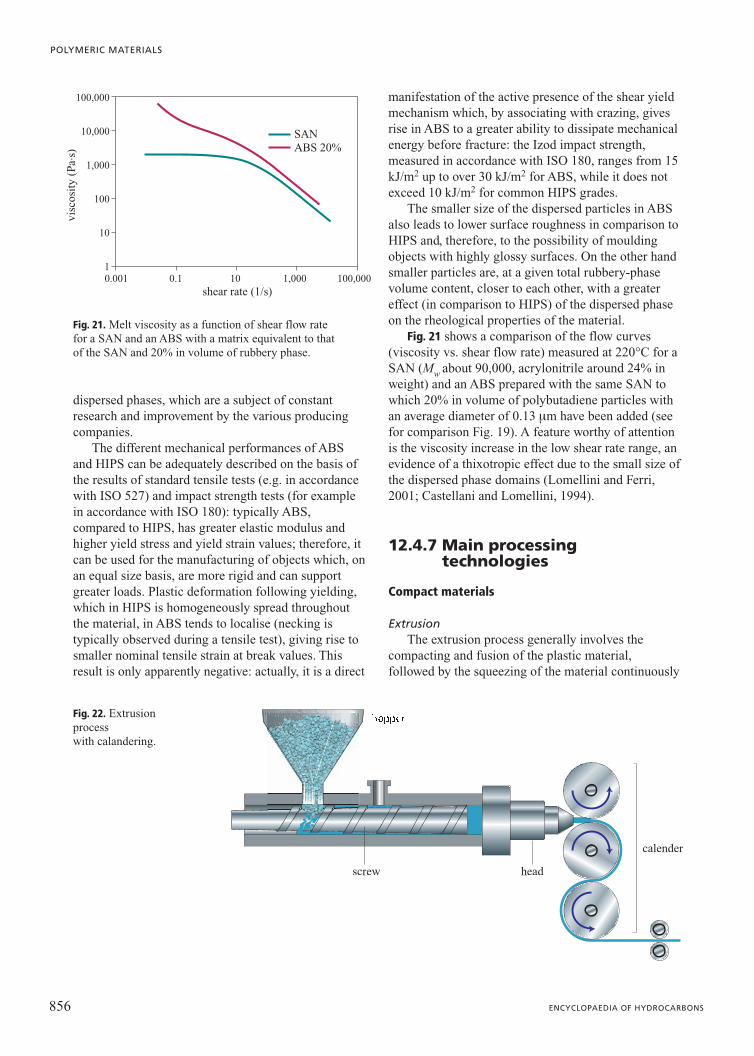
Fig. 21 shows a comparison of the flow curves(viscosity vs. shear flow rate) measured at 220°C for aSAN (Mw about 90,000, acrylonitrile around 24% inweight) and an ABS prepared with the same SAN towhich 20% in volume of polybutadiene particles withan average diameter of 0.13 mm have been added (seefor comparison Fig. 19). A feature worthy of attentionis the viscosity increase in the low shear rate range, anevidence of a thixotropic effect due to the small size ofthe dispersed phase domains (Lomellini and Ferri,2001; Castellani and Lomellini, 1994).
12.4.7 Main processingtechnologies
Compact materials
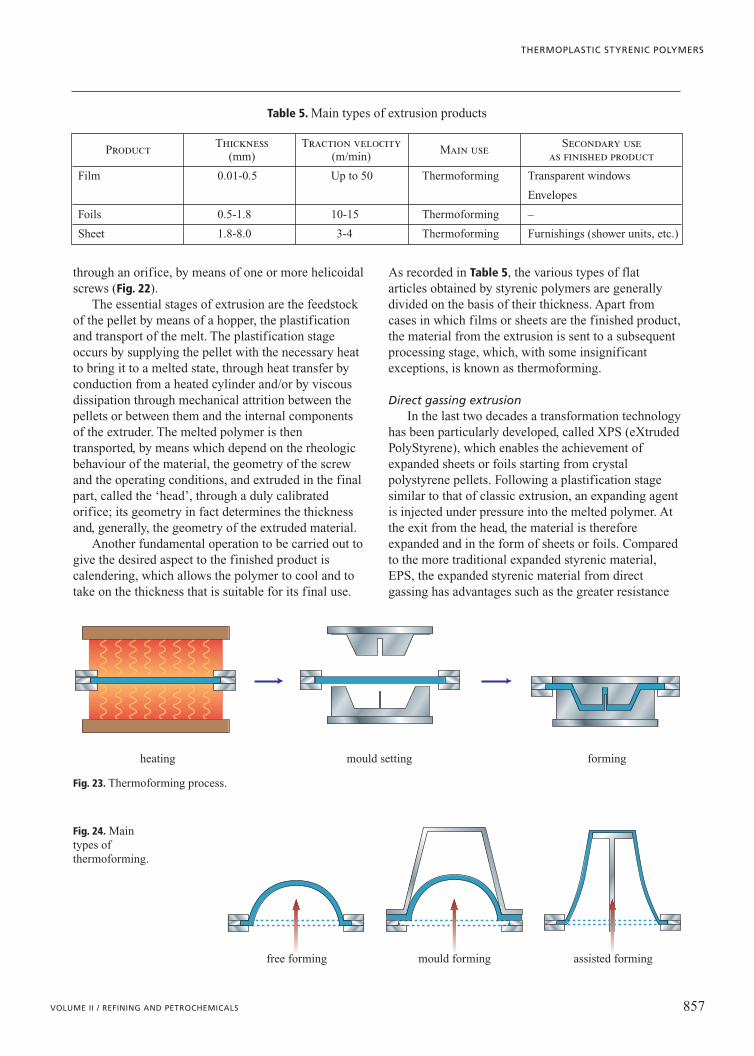
ExtrusionThe extrusion process generally involves the
compacting and fusion of the plastic material,followed by the squeezing of the material continuously
856 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
visc
osit
y (P
a.s)
1
10
100
1,000
10,000
100,000
shear rate (1/s)
SANABS 20%
0.001 0.1 10 1,000 100,000
Fig. 21. Melt viscosity as a function of shear flow rate for a SAN and an ABS with a matrix equivalent to that of the SAN and 20% in volume of rubbery phase.
hopper
screw head
calender
Fig. 22. Extrusionprocess with calandering.
through an orifice, by means of one or more helicoidalscrews (Fig. 22).
The essential stages of extrusion are the feedstockof the pellet by means of a hopper, the plastificationand transport of the melt. The plastification stageoccurs by supplying the pellet with the necessary heatto bring it to a melted state, through heat transfer byconduction from a heated cylinder and/or by viscousdissipation through mechanical attrition between thepellets or between them and the internal componentsof the extruder. The melted polymer is thentransported, by means which depend on the rheologicbehaviour of the material, the geometry of the screwand the operating conditions, and extruded in the finalpart, called the ‘head’, through a duly calibratedorifice; its geometry in fact determines the thicknessand, generally, the geometry of the extruded material.
Another fundamental operation to be carried out togive the desired aspect to the finished product iscalendering, which allows the polymer to cool and totake on the thickness that is suitable for its final use.
As recorded in Table 5, the various types of flatarticles obtained by styrenic polymers are generallydivided on the basis of their thickness. Apart fromcases in which films or sheets are the finished product,the material from the extrusion is sent to a subsequentprocessing stage, which, with some insignificantexceptions, is known as thermoforming.
Direct gassing extrusionIn the last two decades a transformation technology
has been particularly developed, called XPS (eXtrudedPolyStyrene), which enables the achievement ofexpanded sheets or foils starting from crystalpolystyrene pellets. Following a plastification stagesimilar to that of classic extrusion, an expanding agentis injected under pressure into the melted polymer. Atthe exit from the head, the material is thereforeexpanded and in the form of sheets or foils. Comparedto the more traditional expanded styrenic material,EPS, the expanded styrenic material from directgassing has advantages such as the greater resistance
857VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
heating mould setting forming
Fig. 23. Thermoforming process.
free forming mould forming assisted forming
Fig. 24. Maintypes ofthermoforming.
ProductThickness Traction velocity
Main useSecondary use
(mm) (m/min) as finished product
Film 0.01-0.5 Up to 50 Thermoforming Transparent windows
Envelopes
Foils 0.5-1.8 10-15 Thermoforming –
Sheet 1.8-8.0 3-4 Thermoforming Furnishings (shower units, etc.)
Table 5. Main types of extrusion products

to compression and the reduced tendency to absorbwater, while it is penalised in terms of thehomogeneity and uniformity of the cellular structure.Nonetheless, this technology is widely used in theproduction of isolating sheets for construction andfood packaging, typically trays for meat and fish.
ThermoformingThe sheet or foil, obtained by extrusion, is
generally destined for subsequent processing, in whichthe technology known as thermoforming gives thematerial a wide range of shapes.
The process is based on two fundamental stages:heating and forming (Fig. 23).
In the first, the product is inserted in a clampingsystem and heated (using radiant panels or by contact)until the temperature is just above that of the glasstransition (Tg). When the mechanical properties of theproduct are particularly important, it is normal to use arelatively low temperature, thus promoting a greaterdegree of orientation of the material. Where, on theother hand, it is necessary to reproduce particularlycomplicated moulds, the use of higher temperaturesprevails.
The forming phase can be conducted in variousways (Fig. 24). When the product, following heating, isdeformed with a pressurised air jet, free formingoccurs through the application of pressure. Theabsence of contact with mechanical parts gives rise, onthe one hand, to surfaces that are aestheticallysuperior, on the other, to greater problems incontrolling the shape.
Another possibility is that of using a plug (assistedforming), which forces the product into the concavepart of the mould where the application of a vacuumor pressure enables excellent adhesion of the polymerto the walls.
In mould forming, on the other hand, the plasticmaterial adheres to the walls of the mould through theapplication of pressure on the external face of thesheet or of a vacuum on the internal face.
Thermoforming plays a crucial role in the
production of a range of products, from glasses torefrigeration cells.
Injection mouldingThe technology of injection moulding is based on a
peculiar characteristic of the thermoplastic polymers.Once heated beyond their Tg, they can rapidly fill amould, even if this has a complex geometry; followingcooling and, therefore, solidification of the plasticmaterial, the product with the form of the mould canbe extracted and used (Fig. 25).
The initial part of the process in which theplastification of the granules takes place, is not verydifferent from that described for extrusion. However,unlike with extrusion, the plastification screw is notfixed but draws back under the pressure of the flow ofthe polymer during plastification. At the end of thisphase the screw, after suspending the rotarymovement, moves quickly in the opposite direction,pushing the melted polymer into the mould in anaction similar to that of a piston.
A peculiarity of injection moulding is the sectioncommonly called clamping unit, which includes afixed item, where the polymer is directed in channelstowards the mould, and a mobile item, which allowsthe opening of the mould. Following injection and thesubsequent phases of pressurisation and cooling, theproduct can be extracted from the mould.
Compared to thermoforming, injection mouldingallows notably shorter cycle times and the creation ofproducts with more complicated and thickergeometries. The main disadvantages are the investmentcosts linked to the design and construction of themoulds and the difficulty in undertaking large-scalemoulding. Injection moulding generally is not suitablefor the use of polymers with very low fluidity; the highviscosity of the material generates an increase inenergy costs owing to the greater force needed to closethe mould. To obtain a suitable short cycle time, itwould be necessary to use even higher temperatures,with a consequent negative impact on the energyaspects and on the degradation of the polymer.
858 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
hopper
mould
mobileitem
fixeditem
ram-fed injection unitinjection unitclamping unit
Fig. 25. Injectionmoulding process.

Expandable materialsThe suspension process, as already seen, enables
the achievement of beads, of variable diametercontaining from 3 to 7% of expanding agent. Thetransformation of the beads into the finished producttypically takes place in three main phases: expansion(or pre-expansion), ageing, and moulding.
ExpansionDuring the first phase, called expansion (or pre-
expansion), the material reduces its own density until itreaches that of the final application. Inside suitableequipment, the beads are heated to a temperature ofaround 100°C, therefore higher than both the Tg of thepolymer and the boiling point of the expanding agent.The boiling of the expanding agent thus generatespressure within the PS bead, leading to an increase in itsvolume. From the technological viewpoint, the
pre-expansion process can be conducted bothcontinuously and discontinuously (Fig. 26). Continuouspre-expanders allow notable specific productivity, but onthe other hand they are generally less efficient incontrolling density. Modern discontinuous pre-expanders,although less effective in terms of productivity, on theother hand usually drive to an excellent density control.
AgeingAt the end of the expansion, the product is cooled
to the room temperature. The expanding agent and thegas within the cells are, therefore, subject to a markedfall in pressure linked to their condensation. In thisphase the beads are extremely unstable: a lightexternal pressure is enough to deform thempermanently. To balance the internal pressure throughthe diffusion of air within the bead, it is, therefore,necessary to conserve the expanded beads for a fewhours in a silo with permeable walls.
The next step, moulding, requires the presence of aquantity of residual pentane, which depends on thedensity, the type of material and the environmentalconditions. Besides the air permeation, during ageinga further loss of the expanding agent takes also place.For this reason, the duration of the process cannot beextended indefinitely.
859VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS
continuouspre-expander
stirrer
rawmaterial
raw material
weighting cell
levelsensor
stirrer
Archimede’sscrew
wateroutlet
vapourinlet
expandedbeadsoutlet
expandedbeadsoutlet
discontinuouspre-expander
Fig. 26. Continuous and discontinuous pre-expansion process(expandable materials).
filling steaming cooling andblock extraction
Fig. 27. Mouldingprocess (expandable materials).
PolymerEuropean World
market market
SAN 2% 3%
ABS 24% 15%
GPPS/HIPS 57% 58%
EPS 17% 24%
Table 6. Market for thermoplastic styrenic polymers

MouldingThe moulding of EPS consists of a further
expansion in a defined volume: the mould is filledwith pre-expanded and aged beads and then steamheated (Fig. 27). The expanding agent, which asalready seen is still present in the beads, causes afurther expansion of the material within the mould.The pressure generated by the expanding agent, inaddition, compresses the beads against each othercausing their adhesion and therefore synterisation;the degree of adhesion among the beads is calledthe level of synterisation. At the end of moulding,the product is cooled and subsequently subject to afurther stage of ageing. Generally, the beads with amedium to high diameter are moulded in large-scale machines, called block makers, fromwhich blocks are obtained ready to be cut intosheets. The fractions of smaller dimensions arenormally processed into moulds having the finalproduct shape.
12.4.8 The market
Breakdown of styrenic polymers in the world and in Europe
Thermoplastic styrenic polymers have animportant role in the global market formacromolecular materials. In fact, around 12% ofglobal plastic production can be accredited to thiscategory. Table 6 shows the breakdown of productionamong the various types (SAN, ABS, GPPS/HIPS,EPS) with reference to the world and the Europeanmarkets.
Main sectors for application of styrenic polymersThermoplastic styrenic polymers have general
characteristics, such as easy processability, good sizeand appearance stability and balance of mechanicalproperties, which make them particularly suitable for avast range of applications. Following are the mostimportant sectors for their employ, divided by the typeof polymer used.
GPPS/HIPS. The main applications of HIPS andGPPS are: a) packaging: disposable goods (tubs,glasses, etc.), trays, containers for ice cream andyogurt, trays for meat, boxes for sweets and cosmetics;b) electric/electronic: computer and televisionhousings, housings and covers for audio and videocassettes, floppy disks, compact discs, CD-ROMs andDVDs; c) household white goods: cell and internalcomponents of refrigerators and freezers, plastic partsof TVs, computers, washing machines; d) building:insulating panels; e) other: profiles and componentsfor furniture, containers and various articles forkitchens, shower units, laboratory articles, stationery,medical items, toys. Table 7 shows how the Europeanmarket for GPPS and HIPS is divided among theseapplications.
EPS. The main applications for EPS are: building,with insulating panels, formworks for reinforcedcement, lightening for load-bearing structures;packaging, with containers for hot and cold food,protective sleeves; other, with sports helmets, inside ofmotorcycle crash helmets, disposable cores for fusionof metals. Table 8 shows how the European market isdivided among these applications.
ABS/SAN. The application of copolymers based onacrylonitrile, shown in Table 9, is generally similar tothat previously mentioned for GPPS/HIPS. Theadvantage in terms of performance, given the presenceof acrylonitrile, however, allows their use also in othermore specific applications: car interiors, motorbikeparts, lights and other external components, batteryhousings, instrument panels; lighting techniques,windows for caravans, lighters, containers for staticbatteries, bathroom furnishings.
860 ENCYCLOPAEDIA OF HYDROCARBONS
POLYMERIC MATERIALS
Sector Share
Packaging 37%
Electric/electronic 13%
White goods 12%
Building 11%
Other (furniture, household goods, stationery, etc.)
27%
Table 7. Breakdown by applications of GPPS/HIPS
Sector Share
Insulation and building 70%
Packaging 25%
Other 5%
Table 8. Breakdown by applications of EPS
Sector Share
Small household electrical appliances 22%
Transport 19%
Extrusion 16%
Compounds 15%
Electrical/electronic 11%
Other 17%
Table 9. Breakdown by applications of ABS/SAN

References
Amos J.L. (1974) SPE international award address, 1973.Development of impact polystyrene. Review, «PolymerEngineering and Science», 14, 1-11.
Amos J.L. et al. (1954) US Patent 2694692 to Dow.Brandrup J., Immergut E.H. (edited by) (1989) Polymer
handbook, New York, John Wiley.Bucknall C.B. (1977) Toughened plastics, London, Applied
Science.Bucknall C.B. et al. (1989) Rubber toughening of plastics.
Part 12: Deformation mechanisms in toughened Nylon 6.6,«Journal of Materials Science», 24, 2255-2261.
Campbell J.D. et al. (2003) High temperature free radicalpolymerization. Investigation of continuous styrenepolymerisation, «Macromolecules», 36, 5491-5501.
Castellani L., Lomellini P. (1994) Phase volume and sizeeffects on the terminal relaxation of ABS melts, «RheologicaActa», 33, 446-453.
Cigna G. (1970) Dynamic mechanical properties, structure,and composition of impact polystyrene, «Journal of AppliedPolymer Science», 14, 1781-1793.
Claudy P. et al. (1983) Glass transition of polystyrene versusmolecular weight, «Polymer Bulletin», 9, 208-215.
Doi M., Edwards S.F. (1988) The theory of polymer dynamics,Oxford, Oxford University Press.
Donald A.M. (1994) Failure mechanisms in polymericmaterials, in: Collyer A.A. (editor) Rubber toughenedengineering plastics, London, Chapman & Hall, 1-28.
Donald A.M., Kramer E.J. (1982a) Craze initiation andgrowth in high-impact polystyrene, «Journal of AppliedPolymer Science», 27, 3279.
Donald A.M., Kramer E.J. (1982b) Effect of molecularentanglements on craze microstructure, «Journal of PolymerScience. Polymer Physics Edition», 20, 899-909.
Ferrando A., Longo A. (1997) Determination of reactivityratios from 13C NMR analysis of triads fractions for styrene-acrylonitrile copolymerisation as a function of temperature,«Polymer Preprints», 38, 798-799.
Ferry J.D. (1980) Viscoelastic properties of polymers, NewYork, John Wiley.
Georges M.K. et al. (1993) Narrow molecular weight resinsby a free-radical polymerization process, «Macromolecules»,26, 2987-2988.
Goodier J.N. (1933) Concentration of stress around sphericaland cylindrical inclusions and flaws, «Journal of AppliedMechanics», 55, 39.
Hauss A. (1969) Property characteristics of thermally producedpolystyrenes of different molecular weight distribution,«Die Angewandte Makromolekulare Chemie», 8, 73-86.
Haward R.N., Young R.J. (edited by) (1997) The physics ofglassy polymers, London, Chapman & Hall.
Huang N.J., Sundberg D.C. (1995) Fundamental studies ofgrafting reactions in free radical copolymerization. IV:Grafting of styrene, acrylate, and methacrylate monomersonto vinyl-polybutadiene using benzoyl peroxide and AIBNinitiators in solution polymerisation, «Journal of PolymerScience. Part A: Polymer Chemistry», 33, 2587-2603.
Ito K., Yamashita Y. (1965) Copolymer composition andmicrostructure, «Journal of Polymer Science. Part A:Polymer Chemistry», 3, 2165-2187.
Kramer E.J. (1983) Microscopic and molecular fundamentalsof crazing, «Advances in Polymer Science», 52/53, 1-56.
Kramer E.J., Berger L.L. (1990) Fundamental processes ofcraze growth and failure, «Advances in Polymer Science»,91/92, 1-68.
Krevelen D.W. van (1990) Properties of polymers,Amsterdam, Elsevier.
Lazzeri A., Bucknall C.B. (1995) Applications of a dilationalyielding model to rubber-toughened polymers, «Polymer»,36, 2895-2902.
Lomellini P. (1992a) Williams-Landel-Ferry versus Arrheniusbehaviour. Polystyrene melt viscoelasticity revised,«Polymer», 33, 4983-4989.
Lomellini P. (1992b) Effect of chain length on the networkmodulus and entanglement, «Polymer», 33, 1255-1260.
Lomellini P., Ferri D. (2001) Effetto del contenuto di gommasulla reologia estensionale di polimeri tenacizzati conelastomeri, in: Atti del 7° Convegno nazionale di reologiaapplicata, Faenza, 20-22 giugno.
Lomellini P., Rossi A.G. (1990) Effect of composition on theentanglement density in random copolymers, «MakromolareChemie», 191, 1729-1737.
McCormick H.W. et al. (1959) The effect of molecular-weightdistribution on the physical properties of polystyrene,«Journal of Polymer Science», 34, 87-100.
Maestrini C. et al. (1992) Particle entrapping by crazes inHIPS (high-impact polystyrene), «Polymer», 33, 1556-1558.
Matyjaszewsky K., Wang J.S. (1995) Controlled/’living’radical polymerization. Atom transfer radical polymerizationin the presence of transition-metal complexes, «Journal ofthe American Chemical Society», 117, 5614-5615.
Mayo F.R. (1968) The dimerization of styrene, «Journal of theAmerican Chemical Society», 90, 1289-1295.
Olaj O.F. (1971) Zur Kinetik der spontanen und der durchextrem kleine Starterkonzentrationen initiierten radicalischenPolymerisation des Styrols, «Monatshefte für Chemie»,102, 648-671.
Ramsteiner F., Heckmann W. (1985) Mode of deformationin rubber-modified polyamide, «Polymer Communication»,26, 199-200.
Rizzardo E. et al. (1999) Tailored polymers by free radicalprocesses, «Macromolecular Symposia», 143, 291-307.
Rossi A.G. (1984) Relazione tecnica 40/84, Polimeri Europa,Mantova.
Scheirs J., Priddy D.B. (edited by) (2003) Modern styrenicpolymers. Polystyrenes and styrenic copolymers, Chichester,John Wiley.
Staudinger H. (1932) Die hochmolekularen organischenVerbidungen. Kautschuk und Cellulose, Berlin, Springer.
Tsenoglou C. (2000) Molecular rheology of the non-newtonianbehaviour in polymer melts and concentrated solutions, in:Proceedings of the 13th International congress on rheology,Cambridge, 20-25 August, 50-52.
Walker I., Collyer A.A. (1994) Rubber toughenedmechanisms in polymeric materials, in: Collier A.A. (editor)Rubber toughened engineering plastics, London, Chapman& Hall.
Leonardo Castellani Aldo Longo
Francesco Pasquali
Polimeri EuropaMantova, Italy
861VOLUME II / REFINING AND PETROCHEMICALS
THERMOPLASTIC STYRENIC POLYMERS






























