Embed Size (px)
Citation preview

CHAPTER 1
The Occurrence, Dissolution, and Deposition of Silica
Silica is by far the major component of the earth’s crust, yet much remains to be learned of its chemistry and, in particular, its solubility behavior in water. The man- ner of its deposition to form such curiosities as quartz crystals containing inclusions of mineral oil, mercury, or liquid carbon dioxide remains a mystery ( I ) . Flint, which our remote ancestors recognized a s the strongest and toughest stone available, was apparently formed in some instances from the siliceous skeletons of ancient sponges by a mysterious process of solution transport. Within some plants and marine organisms, soluble silica is transported and deposited i n characteristic intricate pat- terns. Only recently has it been recognized that soluble silica, even in trace amounts, plays a role i n the development of mammals.
THE SILICA-WATER SYSTEM
As water is a unique liquid, so is amorphous silica a unique solid. They are much alike, both consisting mainly of oxygen atoms with the smaller hydrogen or silicon atoms in the interstices. As pointed out by Weyl and Marboe (2), “Some properties of water and silica are so similar that the transition between hydrated silicic acids and the aqueous matrix is a gradual one.” Washburn (3) noted that water and amor- phous silica both have a temperature of minimum volume. Ephraim (4) observed another similarity between silica and water in that water is much less dense than expected from close packing of the constituent atoms and from X-ray diffraction studies. Bernal and Fowler (5a) concluded that water molecules are arranged in a rather open structure like quartz, and undercooled water has a still more open struc- ture, like tridymite. Another model has been proposed by Weres and Rice (5b).
These ideas lead to the suggestion that there is some relationship between the density of water and the solubility of the various forms of silica, since both are related to the close packing of oxygen atoms. Both silica and water consist, from the standpoint of volume, largely of oxygen atoms, which are packed together with a characteristic packing density. The small hydrogen and silicon atoms tit between the
3

4 The Occurrence, Dissolution, and Deposition of Silica
oxygen atoms, contributing little to the volume. In pure orthosilicic acid, Si(OH), (if it could be prepared), the small silicon and hydrogen atoms, lying i n the interstices between the large oxygen atoms, would be more or less evenly distributed throughout the mass. Polymerization of silicic acid to form solid silica and water amounts to separation into two phases: in silica, the silicon atoms surround themselves with oxygen atoms in a region of closer packing, and in water hydrogen atoms surround themselves with oxygen in a region of more open packing. In amor- phous S i02 , there are 1.17 grams of oxygen per cubic centimeter; in water of density 1.0, there is 0.89 gram of oxygen per cubic centimeter.
There is no evidence that silica is “soluble” to any appreciable degree in any liquid other than water. However, that statement may depend on the definition of “soluble.” The dissolution of silica involves a chemical reaction or hydrolysis in an excess of water:
SiO, + 2 H,O = %(OH),
Thus i t is not a simple solution such as that of sugar in water, where the sugar molecule exists intact in solution as i n the crystalline state. Instead, it is analogous to a hypothetical equilibrium of silica and ether in an excess of ether:
SiO, + 2 (CH3),0 = Si(OCH,),
Since low condensation polymers such as [(HO),SiO], appear to be clear water- miscible fluids resembling a polyhydroxy organic compound like glycerol (6), the monomer, “soluble silica” or Si(OH),, would probably be a clear liquid if it could be isolated in anhydrous condition. In a pure state i t might even crystallize.
The unusual nature of the silica-water system has been noted by J . A. Kitchener (7) , who pointed out that the endless confusion in the literature concerning the silica-water interface has arisen because the hydration and solubility characteristics have not been understood. For example, there is the question as to why silica sols are extraordinarily stable a t pH 2 where the zeta potential is zero and become increasingly sensitive to electrolytes at higher pH, where the potential is highest-in contradiction to the generally accepted electrical double layer theory. Another mystery is that crystalline quartz becomes coated with a film of amorphous silica even though the solution is undersaturated with soluble silica with respect to a sur- face of amorphous silica.
The dissolution and deposition of silica i n water involves hydration and dehydra- tion reactions catalyzed by O H - ions:
hydration
dehydration (SO,), + 2 H,O (SiO,)x-i + %(OH),
For massive amorphous silica, the equilibrium concentration of Si(OH), at 25OC corresponds to 70 ppm as S O , . This is the “solubility” of anhydrous nonporous amorphous SiO,. However, except for fused SiO, glass, the common forms of amor-

The Silica-Water System 5
phous silica consist of extremely small particles of amorphous silica, or porous aggregates, the surface of which is hydrated as S iOH groups. These exhibit a somewhat higher solubility so that most powders and gels have a solubility of 100- I30 ppm S O , .
O n the other hand, crystalline silica, such a s quartz, almost universally present as “sand.” has a much lower solubility, of the order of 6 ppm SiO,.
Supersaturated solutions of monomeric .%(OH), are formed when silica is dissolved in water a t high temperature under pressure and then cooled, or when an aqueous solution of soluble silicate is acidified:
Na,SiO, + H,O + 2 HCI = Si(OH), + 2 NaCl
Supersaturated solutions of silicic acid in pure water are thermodynamically unsta- ble because condensation polymerization through dehydration takes place. All higher polymers of whatever size, molecular weight, or state of hydration can be represented by a general formula containing n silicon atoms. The polymerization of additional monomer molecules or the deposition of silica can be represented as follows:
where n = number of silicon atoms in a polysilicic acid molecule or particle or polymeric network
x = number of O H groups per silicon atom in the polymer, not exceeding 4 m = the number of monomeric silicic acid molecules added to the polymer p = fraction of the hydroxyl groups per monomeric silicic acid molecule that
are converted to water during the polymerization reaction
Thus when p = I , the monomer is converted to S O , within the polymer molecule without change in the number of O H groups in the polymer. There are, of course, restrictions such as n and m having to be integers and the values of x and p being limited by the possible structures of polymers and conditions of polymerization.
However, for the case where dense amorphous silica is being deposited on exten- sive, massive silica surfaces from slightly supersaturated monomer solution, espe- cially a t high temperature and neutral or alkaline pH, x is very small, p is unity, and n is large. Thus the deposited silica may be essentially dense and anhydrous:
Si,Ozn + m Si(OH), = Sin+mOzn+zm + 2m H,O
Even vitreous or glassy silica contains some water, probably as S iOH groups. At a given temperature and humidity there is an equilibrium “solubility” of water in vit- reous silica, according to Hetherington and Jack (8). Flame-fused quartz contains 0.04 wt. % OH, whereas electrically fused material contains only 0.0003% as detected by infrared absorption a t 2.73 micron wavelength. By extrapolation to

6 The Occurrence, Dissolution, and Deposition of Silica
30°C, Moulson and Roberts (9) concluded the equilibrium concentration of water in silica glass may be a s high a s 0.22% H,O, probably present as internal S iOH groups.
Let us return to the behavior of soluble silica in water. When the solution is highly supersaturated and insufficient solid silica surface is available to permit rapid deposition of soluble silica, new small nuclei particles are formed by intercondensa- tion on monomer and low polymers. Silica is also deposited on these until supersatu- ration is relieved.
I t is in this manner that colloidal particles of silica are formed. These, in turn, may be aggregated to form silica gel or may be laid down as opal, both of which are highly porous wi th an extensive internal surface covered with SiOH groups. Thus "hydrated" silicas are formed. Very slow deposition may produce quartz.
Thermodynamics of the System
The heat of formation of silica by the reaction
Si(c) + 0, (g) = SiO, (s)
was reported ( I O , 1 1 ) : AH:98oK = -217.5 * 0.5 kcal mole- ' for alpha quartz, and -215.9 + 0.3 for amorphous silica.
Greenberg and Price (12) give somewhat different estimated values:
H f Z 8 8 0 K AF (cal mole- ') (cal mole- ')
Alpha quartz (4) -210, 260 Colloidal silica (cs) -205, 570 Vitreous silica (vs)
- 1220 (AFQ - AF,,, 200OC) -550 (AFvs - AF,,, 25OC)
for the overall equilibrium
SiO,(s) + m H,O(I) = H,SiO,(aq)
Greenberg ( 1 3) calculated the following values for the thermodynamic functions:
Amorphous Silica Quartz
AH (kcal mole- ') +2.65 A 0.28 +7.34 * 0.37 AF;980K (kcal mole- ') +3.98 A 0.04 +5.20 += 0.04 L!T&K (cal deg- ' mole- ') -2.82 + 0.50 +4.53 2 0.71
According to these data , the heat of formation of quartz from amorphous solid silica is A H = -4.69 kcal mole- ' , which is greater than the value - 1.78 found by Wise et

The Silica-Water System 7
al. (14). The latter is closer to the value 0.54 * 0.2 more recently calculated by Cochran and Foster (15).
Other reported values for the above hydration reaction of amorphous silica were given by Morey, Fournier, and Rowe ( l6) , who found
AH2e80K = 3 . 3 5 kcal mole-' rind 3FZe8oK = 3.70 kcal mole-'
Kitahara (17a) measured the solubility of amorphous silica between 0 and 100°C ahd calculated A H 2 9 B o K = 3.2 kcal mole-'.
Walther and Helgeson ( l7b) calculated the thermodynamic properties of aqueous silica and the solubility of quartz and its polymorphs over a wide range of tempera- tures and pressures. The thermodynamic constants derived from all available data were evaluated as follows:
Constant Alpha Quartz Amorphous Silica
Entropy, S o (cal deg-I mole-') 9.88 14.34 Volume, V" (cm3 mole-') 22.69 29.0 Gibbs free energy, A G " (cal mole-') - 202.89 Enthalpy, Mf (cal mole-') -2 1 7.65 -214.57
- 204.65
Coefficients were also given with equations for calculating the values over a wide range of temperatures and pressures.
Relating Particle Size and Composition
I n most sols that consist of discrete spherical particles of amorphous silica, the interior of the particles consists of anhydrous SiO, with a density of 2 . 2 g The silicon atoms located at the surface bear O H groups which are not lost when the silica is dried to remove free water.
The relation of particle composition to particle size can be calculated purely from geometry and densities of the components. Let
n, = total number of silicon atoms in a particle n, = number of silicon atoms a t the particle surface d = diameter of particle on anhydrous basis (nm) dh = diameter of hydroxylated particle (nm) x = ratio of SiOH groups to total Si atoms
= n,/n, assuming one O H per surface silicon w = weight of one anhydrous 90, particle (grams) wh = weight of one surface hydroxylated particle p = average number of silicon atoms across the diameter of a particle

8 The Occurrence, Dissolution, and Deposition of Silica
The following equations are pertinent:
7r n , = - p 3
6
or
n, = I1.5dJ = 0.524~~
p = 2.80d = 1.24n:
n, = - [p3 - (p - 2)3] 7r
6
x = 2.14d-I - 1.53d-' + 0.36d-'
The hydroxyl content, expressed as wt. % water, is
'70 H,O = 32d-' - 23d-' + 5 . 4 W 3
The composition of the particle may be expressed as
where 2a t b = 4n. Then in terms o f anhydrous particle diameter (nanometers) the composition is given by
n = 11.5d3
u = 23d3 - 12.3d2 + 8.8d - 2.09
b = 24.6d2 - l7.6d + 4.18
The concentration of hydroxyl groups per square nanometer can be calculated, as well as the OH :Si and 0: Si ratios:
n d d* OH :Si 0 : s ; OH nm-*
8 0.89 I .O 0.99 1.51 2.9 40 I .52 1 . 7 0.85 I .57 4.3
100 2.06 2.2 0.72 I .64 5.0 31 1 3 .O 3 . 2 0.55 1.72 5.7
1438 5 .o 5 . 2 0.37 I .82 6.5 1 1500 10.0 10.2 0.20 I .90 7.2
X X X 0 2.0 7 .8
As discussed in Chapter 6, i n an amorphous structure of this type some of the silicon atoms at the surface are above and some below the average spherical surface, and

The Silica-Water System 9
the number of O H nm-z on the outer silicon atoms is closer to 4.6. However, on very small particles, for example, less than 5 nm in diameter, the value is probably closer to the above calculated values.
Energy Change with Changing Particle Size and Composition
The change in free energy with increase in particle size must be quite small. Let us consider a number of particles of silica with silanol surfaces suspended in water, which are to be converted to one larger particle with loss of surface area. The com- position of the silica surface will remain unchanged. The following steps are dis- cussed only from the standpoint of the approximate changes in free energy. Only heat effects can be considered because the entropy changes are not known, but they are assumed to be small or t o cancel out, and so can be neglected.
I . Adsorbed water is removed from the area of the silanol surface that is to be diminished. This requires an input of 46 ca1/1000 mz or 190 ergs cm-2 (18) cor- responding to the heat of wetting.
2. The silanol surface is fully dehydrated to a siloxane surface (19). This requires an input of 4660 230 cal mole-' of water removed.
3. The siloxane surfaces are brought together to reduce the surface area. This results in an evolution of heat equal to the loss of surface energy which is 259 f 3 ergs cm -z (19), or 63 cal per 1000 m2 for a siloxane surface.
From this the overall energy change can be calculated for a decrease iin surface area of 1000 m2 g- ' of silica. I n step 2 i f i t is assumed there are 8 O H or 4 H 2 0 nm-2 to be evolved, this would require 31 cal per 1000 m*. Thus the net energy change would be -46 - 31 + 63 or - 14 cal per 1000.m2. On the other hand, if the commonly measured value of 4.6 O H nm-2 is used, requiring 18 cal per 1000 mz for removal, the net energy consumption would be -46 - 18 + 63, or - I cal per 1000 mz.
From the change i n solubility with particle size, the value for the interfacial energy is about 46 cal c r r 2 , corresponding to an evolution of I 1 cal m-*.
Another approach is to consider the energy of the silanol surface of 129 * 8 ergs cm-I (19), corresponding to an evolution of 31 cal per 1000 mz when the surface area decreases. However, the water adsorbed on the silanol surface, corresponding to the heat of wetting, has to be desorbed, consuming 46 cal per 1000 mz. Thus the decrease i n area would require a total consumption of 46 - 31 = 15 cal per 1000 mz.
However, without data on the entropy changes involved, the relation between heat effects and free energy changes cannot be accurately established.
It therefore appears that i f we consider only silica surfaces where the radius of curvature is well above molecular dimensions, there should be little tendency for the area of the silica-water interface to decrease at 25OC.
Regardless of these considerations it is apparent that there is very little change in energy content of aqueous silica sols or gels with change in surface area, once the

10 The Occurrence, Dissolution, and Deposition of Silica
surface had decreased and the particles have grown to a certain size. Further spon- taneous changes are unlikely to occur.
One point that has not been considered i n the foregoing discussion is that the energ) values have been generally determined on types of silicas that have already reached a relativel) stabilized state of particle growth. On the other hand, for much finer silica, for example, with a specific surface of more than 600 m2 g-’, the radius of curvature of the surface is then less than 2 5 A. and the silanol groups must be spread apart so that less hydrogen bonding can occur between neighboring hydroxyl groups. I n turn, it might be expected that this would increase the heat of wetting, decrease the heat of dehydration. and decrease particle density and surface energy. Under these conditions, i t is certain that particle growth occurs with decrease in the radius of curvature, but energy data on such materials have not been obtained, particularly i n regard to surface energy of the silanol-water interface.
SOLUBLE SILICA-MONOSILICIC ACID
The soluble form of silica is monomeric, containing only one silicon atom and generall) formulated as Si(OH),. This is often called monosilicic acid or orthosilicic acid. The state of hydration is not known, although a t high pressure there is some indication that one water molecule is linked to each OH group, probably by hydrogen bonding. so the hydrated molecule is represented by Willey (20) as Si(0H :OH,),.
The structure of monosilicic acid is assumed to involve silicon coordinated with four okygen atoms as in amorphous vitreous silica and i n crystalline quartz. Although there are rare minerals such as the stishovite form of SiO, (21) or thau- masite (22 ) . i n which silicon is coordinated with six oxygen atoms, silicon i n most oxides and silicates is surrounded by only four oxygen atoms. I f the monomer had the structure H,Si(OH),, one would expect i t to be a strong acid like the analogous H,SiF,. but i n fact i t is a very weak acid.
I t is essentially nonionic in neutral and weakly acidic solution and is not transported by electric current unless ionized in alkaline solution. It is not salted out of water nor can it be extracted by neutral organic solvents.
It remains i n the monomeric state for long periods in water a t 25°C. as long as the concentration is less than about 2 x M , but polymerizes, usually rapidly, at higher concentrations, initially forming polysilicic acids of low molecular weight and then larger polymeric species recognizable as colloidal particles.
The question often arises a s to whether the term “soluble silica” should include the I O M pollmers such as tetramer or decamer, which are classed as “oligomers.” I t becomes a matter of definition. ”Soluble” materials have been recognized as those that pass through a dial>sis membrane, whereas colloids d o not; but even though membranes can nou be made bi th pores sufficiently small to separate dextrose from sucrose. we think of sucrose a s being ”soluble.” On the other hand, sucrose is cer- tainly not colloidal.
For the purpose of this book. the following terminolog) is used:

Soluble Silica-Monosilicic Acid I 1
Soluble silica (or monosilicic acid). Si(OH), Polysilicic acid (oligomers). Polymers with molecular weights (as SiO,) up to
about 100,000, whether consisting of highly hydrated “active” silica or dense spherical particles less than about 50 A in diameter.
Colloidal silica. More highly polymerized species or particles larger than about 50 A, although sometimes down to 10-20 A.
Silica sol. May refer broadly either to polysilicic acid or colloidal silica
The arbitrary borderline of 50 A or mol. wt. 100,000 is based on the general observation that below this point the polymer species are generally unstable, having only a transient existence owing to gelling or particle growth.
Also, as has already been shown, it is below this size range that less than half of all the silicon atoms are present a s SiO, that is, as ‘‘silica,” whereas more than half are each associated with at least one hydroxyl group. The term “silicic” acid is thus justifiable.
The preparation and reactions, for example, polymerization, of dilute solutions of monosilicic acid are further described in Chapter 3. Meanwhile, some of its charac- teristics are noted, as follows, prior to discussing solubility:
I . It is characterized by its rapid rate of reaction with molybdic acid to form the yellow silicomolybdic acid.
2 . It is generally inert in neutral solution if the concentration is below the saturation level with respect to amorphous silica. Thus i t is almost universally present at a concentration of a few parts per million in most natural waters and i n living organ isms.
3. I t combines with metal ions to an increasing degree with increasing pH. thus reducing the concentration of free monosilicic acid. (Ferric and uranyl ions react a t a pH as low as 2 , whereas most other metal ions combine only a t higher pH.)
4. Above pH 9 i t is ionized first to (HO),SiO- or at still higher pH to (HO),Si0,2-. The first equilibrium constant (13, 23) is approximately (25°C)
[(HO),SiO-]
[ O H - ] [Si(OH),I = 1.5 x 1 0 4
or
Even though the silica solution is neutral. i f i t is passed through a bed of strong- base cation-exchange resin in the free-base form, the soluble silica in contact with the resin is ionized and is then held as silicate ions. I n a mixture of Si(OH), in equilibrium with colloidal silica particles at pH 7-8, the particles bear a negative charge. According to Goto. Okura, and Kayama (24) electrophoresis and trans-

I2 The Occurrence. Dissolution, and Deposition of Silica
port studies show that the colloid, not the “molecularly soluble” %(OH),, is the charge carrier. When the mixture is passed through a mixture of strong-base anion- and cation-exchange resin, monosilicic acid is removed, but not the colloidal particles. [After a t ime the particles dissolve sufficiently to reestablish the equilibrium concentration of Si(OH),.]
5 . I t is converted to H,SiF, by reaction with H F i n aqueous solution:
%(OH) , + 6 H F = 2 H’ + SiF:- + 4 H,O
6. I t is converted to a complex anion by reaction with o-dihydroxy aromatic com- pounds. such as catechol, i n neutral solution:
Si(OH), + 3 o-C,H,(OH), + 2 NH; + 2 OH- =
(o-C2H401)3Si2- + 2 NH,- + 8 H,O
Volatility in Steam
Although Si(OH), is nonvolatile a t ordinary temperature and polymerizes quickly when heated, nevertheless a t elevated temperature and pressure in water its solubility is greatly increased and it can exist in equilibrium as the vapor phase in the steam, as shown by Kennedy (25). This is of importance in very high pressure boilers in power plants where deposits build up on turbine blades unless all silica is removed from the feedwater. Brady (26) supposes the volatile species is Si(OH), or (HO),SiOSi(OH),. Astrand (27) found that volatility increased with decreasing alkalinity i n experiments conducted u p t o 350°C and 300 atm. This, of course, sug- gests that .%(OH), is more volatile than the silicate ion. Wendlandt and Glemser (28) reviewed evidence from earlier workers and calculated the equilibrium constants involved whence the species i n the vapor were related to the density of the water vapor:
Quartz Gas Density Range (g cm-3)
SiO, + 2 H,O = Si(OH), U p to 0.05 2 SiO, + 3 H,O = (HO),SiOSi(OH), U p to 0.45 SiO, + H,O = OSi(OH), Above 0.65
Similarly, Mart jnova, Fursenko, and Popov (29) found that i n a solution satu- rated with soluble silica at 263-364°C about a third of the silica in the vapor was present a s disilicic acid, whereas i n the range 151-223°C i t was all monomeric.
Heitmann (30) concluded that deposition in turbines was minimal i f the silica concentration was less than 0.01 ppm. iron concentration less than 0.005 ppm, and the conductivity less than 0. I micromho cm-’ . According to Heitmann’s measure- ments (31), the silica concentration in the vapor phase ranges from 0.1 mg kg- ’ a t 400°C to 5 mg kg-’ at 600°C at a pressure of 0.3 kg cm-*, but increases t o more than 100 mg kg- ’ under an applied pressure of 300 kg cm-l .

Soluble Silica-Monosilicic Acid 13
Soluble Silica in Nature
Silica is constantly dissolving and precipitating over a large part of the earth’s sur- face. The sedimentary cycles have been described in complex detail by Siever (32). Soluble silica is mainly derived from the weathering of minerals which, in some cases, results in amorphous silica residues that then dissolve. Very little can come from the “sands of seashore,” or quartz, which is soluble to only a few parts per million; furthermore, the rate of dissolution is extremely slow. River waters range from 5 to 35 ppm SO,, a few up to 75 ppm, and by the time they reach the sea may range from 5 to 15 ppm. Seawater varies widely but the silica content may range from 2 to 14 ppm (33). However, Lisitsyn and Bogdanov (34) report that surface waters in the Pacific Ocean contain only 0.0001-0.3 ppm S O z . Plankton convert 6 x IO8 tons SiOl from soluble to suspended form each year, but this is only 0.16% of the available silica.
In addition to the silica carried into the sea by fresh water, additional soluble silica comes from the suspended colloidal clays and related minerals. Tests show that common colloidal silicates like clay will dissolve i n seawater sufficiently to give a silica concentration of I O ppm (35).
Concentrations of silica of around 2 ppm were reached in dilute salt solution with mica and kaolin and up to 15 ppm with montmorillonite (36). When seawater was enriched with soluble silica to 25 ppm SO,, i t remained at that level for a year in the absence of these minerals, but when the latter were then added, the silica was removed from solution down to the 2-15 ppm level that was reached when the minerals alone were added. Since many ocean waters contain 2-10 ppm SO,, it is possible that this value is reached as the equilibrium solubility of colloidal alumino- silicate in suspension. The above experiment is consistent with the fact that in pure water. pure amorphous silica dissolves to give a concentration of monosilicic acid of 100-110 ppm, but in the presence of polyvalent metal cations such as iron, alu- minum, and other metals, colloidal silicates are formed with a much lower solubility with respect to monosilicic acid. Iler (37) has shown that soluble aluminum reduces the solubility of amorphous silica from about I10 to less than I O ppm.
Willey (38, 39) has studied the natural interaction of soluble alumina and soluble silica in 0.6 N sodium chloride solution. Addition of aluminum ion to 200 ppm Si(OH), retards polymerization. Probably there is formed a colloidal complex which reacts as monomer when put into the strongly acidic molybdate reagent. A very low concentration of soluble silica also causes the precipitation of alumina.
Soluble silica as determined by the molybdate method is not necessarily present as Si(OH),. Bogdanova (40) reported that in natural waters that contained only about 5 ppm total silica, 4 - 9 s of the silica was polymeric but was converted to monomer by acid. It is most likely that the “polymeric” silica was actually very small colloidal particles of aluminum silicate that liberated monomer when acidified.
Silica is continuously removed from seawater by biochemical processes. Diatoms and sponges as well as plants remove silica which is stored within the organism. Although Calvert (41) believes that the concentration of silica in the sea is mainly controlled by biological activity, Harder (42) reports that amorphous hydroxides of AI, Fe, Mn, or Mg can react with and precipitate soluble silica, thus reducing the concentration to as low as 3 ppm. Both processes are no doubt operative.
I4 The Occurrence, Dissolution, and Deposition of Silica
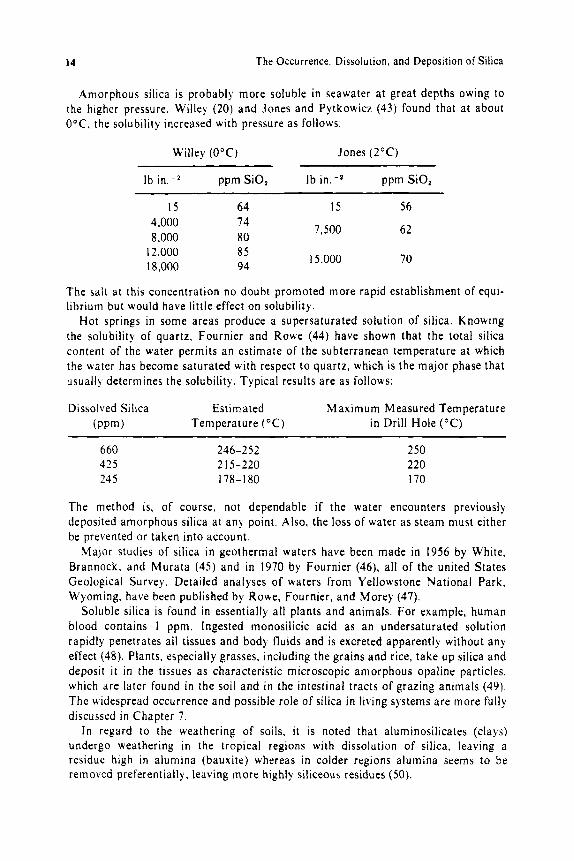
Amorphous silica is probably more soluble in seawater a t great depths owing to the higher pressure. Willey (20) and Jones and Pytkowicz (43) found that at about O°C, the solubility increased with pressure as follows:
Willey ( O O C ) Jones (2OC)
Ib in:* ppm SiO, Ib in.-2 ppm SiO,
15 64 15 56 4,000 8,000
12,000 18,000
74 80 85 94
7,500
15.000
62
70
The salt a t this concentration no doubt promoted more rapid establishment of equi- librium but would have little effect on solubility.
Hot springs in some areas produce a supersaturated solution of silica. Knowing the solubility of quartz, Fournier and Rowe (44) have shown that the total silica content of the water permits an estimate of the subterranean temperature a t which the water has become saturated with respect to quartz, which is the major phase that usually determines the solubility. Typical results are as follows:
Dissolved Silica Estim ated Maximum Measured Temperature (PPm) Temperature ("C) in Drill Hole ("C)
660 246-252 250 425 2 15-220 220 245 178-180 170
The method is, of course, not dependable if the water encounters previously deposited amorphous silica a t any point. Also, the loss of water as steam must either be prevented or taken into account.
Major studies of silica in geothermal waters have been made in 1956 by White, Brannock, and Murata (45) and i n 1970 by Fournier (46), all of the united States Geological Survey. Detailed analyses of waters from Yellowstone National Park, Wyoming, have been published by Rowe, Fournier, and Morey (47).
Soluble silica is found in essentially all plants and animals. For example, human blood contains I ppm. Ingested monosilicic acid as an undersaturated solution rapidly penetrates all tissues and body fluids and is excreted apparently without any effect (48). Plants, especially grasses, including the grains and rice, take up silica and deposit i t i n the tissues as characteristic microscopic amorphous opaline particles, which a re later found i n the soil and i n the intestinal tracts o f grazing animals (49). The widespread occurrence and possible role of silica in living systems are more fully discussed i n Chapter 7.
I n regard to the weathering of soils, i t is noted that aluminosilicates (clays) undergo weathering in the tropical regions with dissolution of silica, leaving a residue high in alumina (bauxite) whereas i n colder regions alumina seems to be removed preferentially, leaving more highly siliceous residues (50).

Phases of Silica 15
A possible explanation is that in the tropics the decomposing vegetation produces tannins and other catechol-like materials that are known to dissolve silica i n neutral solution. I n colder regions, less organic matter is likely to be present and the pH may be lower because more dissolved carbon dioxide is present, so that alumina is preferentially dissolved.
PHASES OF SILICA
Since different phases of silica exhibit different solubility behavior, they are briefly described. By far the commonest crystalline form is quartz, the main constituent of common sand. However, under certain conditions in nature and in the laboratory, other forms a re produced. These forms in t u r n may be divided into the following classes:
I. 2. 3.
4 .
5.
Anhydrous crystalline SO,. Hydrated crystalline SiO, .xH,O. Anhydrous amorphous silica of microporous anisotropic form such as fibers or sheets. Anhydrous and hydrous amorphous silica of colloidally subdivided or microporous isotropic form such as sols, gels, and fine powders. Massive dense amorphous silica glass.
Of these, 2, 3, and 4 exhibit extensive external or internal surfaces and are thus pertinent to the present study.
Anhydrous Crystalline Silicas
Sosman (51) classified the more common phases as follows:
Thermodynamically Stable a t Atmospheric Thermodynamic
Pressure Stability Range ("C)
Quartz low Quartz high Tridyrnite S-l Tridymite S-I1 Tridymite S-111 Tridymite S-IV Tridymite S-V Tridymite S-VI Tridymite M-I Tridyrnite M-II Tridymite M-I11 Cristobalite low Cristobalite high
T o 573 573-867 -, tridymite To 64 64-1 I7 117-163 163-2 I O 210-475 475-1470 -+ cristobalite To 117
Above 163 T o 272
117-163
272-1723

16 The Occurrence. Dissolution. and Deposition of Silica
The different forms of quartz, tridymite, and cristobalite are transformed spon- taneously with temperature so that from the standpoint of solubility there are only the three phases to be considered.
The next group of three phases are those formed only under conditions of high temperature and pressure.
Thermodynamically Stable Range Thermodynamically
Stable at High Pressure Temperature ("C) Pressure (kilobars) ~~ ~
Keatite Coesite
Stishovite
400-500 From 300 To 1700
1200- 1400
0.8-1.3 15 40
160
Survels of these phases and their properties have been published by Frondel ( 5 2 ) , Sosman (53). and Florke (54). Wells relates the structure of the different forms of silica to various crystalline silicates ( 5 5 ) .
Quartz, the commonest phase found in nature, ranges from huge crystals, to amor- phous-looking powders a few microns in size, to shapeless masses of chalcedony agate or flint consisting of densely packed, interlocked microscopic crystals. T h e transformations between the three common forms and vitreous silica is a s follows:
870°C 1470°C I700"C quartz tridymite cristobalite vitreous
The transformation to tridymite apparently requires traces of certain impurities or mineralizers.
The three phases metastable at ordinary pressure were recognized only recently. Keatite was discovered by Paul Keat (56) i n 1954, and its formation via cristobalite and transformation to quartz were studied by Carr and Fyfe ( 5 7 ) . Hoover (58), i n a patent filed i n 1954, described the preparation of a very similar if not identical material from "silicic acid", that is, hydrated amorphous silica powder, by heating it in water at about 3000 atm pressure and 500-625OC i n the presence of about 1 % alkali based on silica.
Coesite was discovered by Coes, in 1953 (59). I t is made from amorphous silica i n the same temperature range as for keatite. but at I O times the pressure and with weakl) acidic cataljsts such as boric acid or ammonium chloride (59). I t was found i n nature in 1960 at Meteor Crater, Arizona, apparently formed under the high temperature and pressure conditions o f the impact.
Similarl). stishovite was first made in the laboratory i n 1961 by Stishov and Popova (60) and discovered in Meteor Crater by Chao. Shoemaker, and Madsen, in 1962 (61). A most interesting story of the isolation of substantial amounts of coesite and stishovitt: from the crater is told by Bohn and Stober (62, 63).
There are also some unusual anhydrous crystalline forms, as follows (64) .

Phases of Silica 17
Silica W is a fibrous crystalline silica with a density of 1.97 g cm-, described by Weiss and Weiss (65), formed in the gas phase by oxidizing silicon monoxide vapor at 1200-1400°C and deposited as paperlike films. It is unstable above about 1400°C. It is fairly stable i n dry air, but is converted by moisture to amorphous hydrated silica, still retaining a swollen fibrous form. I n this transformation, only about 0.08 mole of H,O is taken up per mole of S O , , forming SiOH groups. Silica W can have no true equilibrium solubility i n water. Instead, i t must decompose rapidly i n water to give monosilicic acid. When the powder is suspended i n water and within 2 min centrifuged to obtain a clear solution, then titrated with N a O H solution at pH 10.2-10.5 (thymolphthalein), 2 equivalents of base are required per mole of SiO, in solution (66). After the solution has been aged for I hr, only 0.1 equivalent is required. Initially, therefore, the solution must have been supersaturated with Si(OH),, which when titrated with base, requires 2 equivalents of alkali, but after the monomer has polymerized, much less alkali is required to neutralize the surface acidity of the colloid. If the solution is mixed with silver salt an orange, light-sensi- tive AgSiO, is precipitated. In absolute methanol the fibers swell and form a polymeric methyl ester containing one methoxy group per silicon atom which, when heated in vacuum at 30O-50O0C, yields cyclic methyl esters [(CH,O),SiO], .3.4.6. When hydration o f the fiber by water is followed by suitable technique under the microscope i t can be seen that the reaction starts at the end of the fiber and proceeds rapidly along its length as the crystal swells and is converted to hydrated amorphous silica:
Anhydrous fibrous silicas formed i n connection with high temperature metallur- gical operations were noted as long ago as 1852 by Schnabel and 1859 by Rose. Soft, silky fibers of more than 98% SiO, were classed as aphanitic (invisible) silica, and also known as lussatite. Around 1910, i n the mouths of electric furnaces making silicon carbide, a soft spongy gray deposit called “elephant’s ear” was identified as microfibrous amorphous silica (67). It is likely that all of these were silica W.
Melunoph/ogite, a long known but strange and little understood mineral. is found i n volcanic sulfur deposits in Sicily. Skinner and Appleman reviewed its history and showed that i t was a new cubic polymorph of silica (68). It has a cubic, very open structure, containing 92.4% SiO, and about 5.7% SO, (2.28% as sulfur), 1.2% carbon. and 0.81% hydrogen. The density is 2.052 * 0.013 g crn- , . The initial refractive index is 1.467, but when these volatile materials are driven off by heating, the crystalline silica residue has a refractive index of 1.425 f 0.002 and a density of 1.99 g cm-3, which are substantially lower than those of amorphous or glassy silica. The silica crystal is stable up to about 900”C, above which it changes into cristo- balite. However, when crystals are subjected to grinding in a mortar at ordinary temperature, the open structure collapses to fine-grained quartz. Its solubility has not been measured, and it is doubtful i f it can have a true equilibrium solubility in

16 The Occurrence, Dissolution. and Deposition of Silica
water. The heat-purified material will probably react with water rapidly to give a highly supersaturated solution of Si(OH),, similar to the behavior of silica W.
The nature of the hydrocarbon and sulfur content is still not clear. However, cal- culations based on density data would seem to support earlier suggestions that the sulfur must be present a s SO, or H,SO, within the silica lattice. The optical charac- teristics of the mineral show that the organic matter occurs in films between the faces of the crystals. On the other hand, calculations based on the difference in densities of the original mineral and the pyrolyzed silica crystals show that the sulfur compounds a t least must be within the crystal lattice. Kamb offers evidence (69) that the silica structure is a clathrate with SO,, H,O, and C H I in the lattice analogous to the known 12 %r gas hydrates of water. 6X.46HZO, where X is CH,, H2S, CO,. SOz, CI,, etc., and i n fact the structure is the complete analogue of 6CI,. H,O.
Silica 0 crystallizes from lithium silicate glasses during devitrification at low temperature. I t has a crystal lattice similar to quartz and may simply be “high quartz” stabilized below 573°C. the normal transition temperature, to low quartz (53, 70) by inclusions of metal ion impurities. The only way pure material can be obtained is by neutron bombardment of quartz.
Silica X is a microcrystalline form obtained as spherical aggregates of radial fibers u p to I2 microns in diameter by heating pure amorphous hydrated silica (“silicic acid”) with 2% K O H solution in sealed tubes at 150°C for a few weeks. The refractive index of 1.484 f 0.004 is close to that of cristobalite. I t is anhydrous, maintaining its structure up to about 600°C, above which it is converted to cristo- balite (71a. 71b).
Silicalire. a very unusual new form of anhydrous crystalline silica homogeneously permeated by uniform pores 6 A i n diameter and having a density of only 1.76 g c m 3 . has been described by Flanigen et al. (71c) and patented by Grose and Flanigen (71d). The pores constitute 33% o f the volume of the crystal. A most remarkable feature is that this silica is hydrophobic; the pores are lined with oxygen atoms that are highly hydrophobic and organophilic or oleophilic. Thus the crystals preferentiall). absorb hexane in the presence of water, which does not enter the pores even at saturation pressure.
This type of silica is made first as a crystalline quaternary ammonium silicate, for example. tetrapropylammonium silicate: ( T P A ) , 0 . 4 8 S i 0 2 . H,O. I t is then heated to red heat to remove the organic matter and water. leaving uniform cylindrical chan- nels throughout the three-dimensional crystalline framework of silica.
A similar but even more hydrophobic. anhydrous, microporous crystalline silica was obtained by Flanigen and Patton ( 7 le) by conducting the hydrothermal synthe- sis i n the presence of some ammonium fluoride. which facilitated the formation of crystals 2-15 microns i n size a t only 100DC rather than at the higher temperature and pressure required for making silicalite. After crystallization from solution a ‘!pica1 composition was about 88% b! Height of silica, 11.0% tetrapropylammonium oxide. and 0.9% fluorine. but after calcination at 600°C the porous crystalline product was essentially pure SiO, (containing less than 0.190 fluorine) with a mean refractive index of 1.39 + 0.01 and a specific gravity of 1.70 * 0.05. These values a re the same as those of silicalite and fall on the same curve with other forms of silica i n Figure I . 1 .
Phases of Silica 19
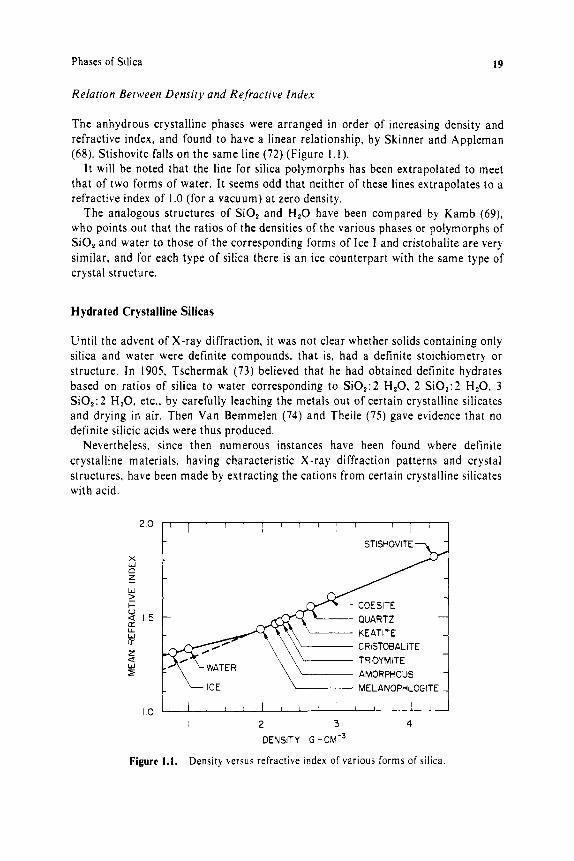
Relation Between Density and Refractive Index
The anhydrous crystalline phases were arranged in order of increasing density and refractive index, and found to have a linear relationship, by Skinner and Appleman (68). Stishovite falls on the same line (72 ) (Figure 1 . 1 ) .
I t will be noted that the line for silica polymorphs has been extrapolated to meet that of two forms of water. It seems odd that neither of these lines extrapolates to a refractive index of 1.0 (for a vacuum) at zero density.
The analogous structures of S O , and H,O have been compared by Kamb (69), who points out that the ratios of the densities of the various phases or polymorphs of SiO, and water to those of the corresponding forms of Ice I and cristobalite are very similar, and for each type of silica there is an ice counterpart with the same type of crystal structure.
Hydrated Crystalline Silicas
Until the advent of X-ray diffraction, i t was not clear whether solids containing only silica and water were definite compounds, that is, had a definite stoichiometry or structure. In 1905. Tschermak (73 ) believed that he had obtained definite hydrates based on ratios of silica to water corresponding to Si0,:2 H,O, 2 Si0, :2 H,O. 3 SiO,: 2 H,O. etc., by carefully leaching the metals out of certain crystalline silicates and drying in air. Then Van Bemmelen ( 7 4 ) and Theile (75 ) gave evidence that no definite silicic acids were thus produced.
Nevertheless, since then numerous instances have been found where definite crystalline materials, having characteristic X-ray diffraction patterns and crystal structures, have been made by extracting the cations from certain crystalline silicates with acid.
2 0 1 1 , I I 1 I I I I , I I 1 I I 1
X W 0 z W 2 I-
% 15 (r L L W U 2 U W 5
I O
CRISTOBALITE
MELANOPHLOGITE -
, 1 1 1 I l l I I I I I
I 2 3 4
DENSITY G - C M - 3
Figure 1.1. Density versus refractive index of various forms of silica.

20 The Occurrence, Dissolution, and Deposition of Silica
There is no instance of a hydrated silica being crystallized directly from a solution of silica and water. Yet there is the peculiarity that certain hydrates, once formed, exhibit what seems to be a characteristic solubility, implying that since an equilib- rium is reached. silica must pass from solution to the solid phase as well as the reverse. However, the data are conflicting.
Sodium disilicafe, Na,Si,O,, may be converted to hydrated silica. Thus by treating the silicate with concentrated cold acid, washing out salts with water and water with acetone, and drying in vacuum at 40”C, a crystalline “disilicic acid” is obtained (76, 77). Such layerlike structures are termed “lepidoidal” (scalelike) or phylloidal (leaf- like), Liebau distinguished two types of layer structures. They are internally hydrogen bonded and exhibit only weak acidity (78).
Crystalline Na$iO,.IH,O gives another form of silica. Here treatment with dry HCI gas at -25OC gave definite silica hydrates containing 2.0, 1.5, and 1.0 moles of H,O per mole of SiO,, as the product was dried under vacuum at progressively increasing temperatures. The silica was anhydrous at 90°C (79).
From gillispire, BaFeSi,O,,, Pabst (80) obtained flakes of crystalline hydrated sil- ica consisting of Si,Olt- ions linked into sheets with the composition 4 H,Si,O,, + 2 H,O: density calculated 2.15. found 2.05; refractive index 1.45 0.01.
From sheerlike KHSi,O,, Le Behan, Kalt, and Wey (81) obtained by treatment with a dilute acid a still different leaflike disilicic acid, H,Si,O,.
From the zeolite mordenite. Chen and Smith (82) obtained silica having the same zeolitic skeleton by repeatedly heating the hydrogen form in steam at 538°C and extracting the liberated aluminum wi th acid. The pores are apparently lined with siloxane oxygen bonds and are hydrophobic, retaining high hydrocarbon sorption capacity.
Chrysotile asbesros, (HO),Mg,.Si,O,, , treated with acid yields fibrous hydrated silica. By reacting the silica with (CH,),SiCI as rapidly as it is liberated, it is converted to the trimethylsilyl derivative which, i n organic solvent, is a highly swollen mass of ribbons of silica only a few angstroms thick. When solvent is removed the ribbons roll up again into tubes or fibers with a structure similar to that of the original fibrous mineral. The fibrous product by X-ray diffraction shows a prominent 15 A d-spacing (83).
Magadiite, NaSi,0,,.3H,O, is a mineral first found in Kenya by Eugster (84) and later i n Oregon and California. However, a similar structure had been synthesized years before by McCulloch (85) by prolonged aging of a mixture of microamorphous silica and alkali (SO,: Na,O = 4-5) in concentrated suspension at IOO°C. ller (86, 87) prepared i t in good yield from a mixture of sodium silicate and colloidal silica (SiO,:Na,O = 4.6) and made a number of derivatives by esterification and ion exchange. McCulloch reported the product to have a SiO,:Na,O ratio of 9.4, and Iler found 8.0, whereas the ratio of magadiite was 14.0. However, ller found his material, when treated with acid, to exchange all the sodium for hydrogen ions, and the crystalline silicic acid had the same X-ray pattern (within +2% d-spacings) as reported by Gude and Sheppard (88) for “silhydrite,” 3SiO,, H,O, which is a crystalline silicic acid found i n association wi th magadiite, The acid obtained by leaching magadiite was reported to be somewhat different. The structure of maga-

Phases of Silica 21
diite has been further studied by Brindley (89), McAtee, House, and Eugster (90). Bricker (91a) measured the stability constant:
[Na*] [H,SiO,]’
[H+I [HzOI’ - 14.3 log10
and also calculated the free energy of formation as AGO = -1762.2 kcal mole-’, where G = standard molal Gibbs free energy.
There is probably a family of similar high-silica crystalline silicates having charac- teristic crystal structures, and from which corresponding crystalline silicic acids could be obtained. Thus, along with magadiite, is found kenyaite, with a SiO,:Na,O ratio of 22; its formula is NaSi,,02,.,(OH),. 3H,O (84). Lagaly and co-workers (91b, 91 c) have further described these “phyllosilicic acids” in regard to structure and ion- exchange properties (see Chapters 2 and 5).
Lepidoidul silica is a term that has been used in the past to describe silica i n the form of extremely thin flakes or scales. Such silicas are usually hydrated and may have a crystalline structure or may be amorphous, depending on how they are made. Three types that have been described are as follows:
I . A synthetic silica [ (s i60E)08](oH)E prepared from siloxane [ HO.Ch-&iH]n
2. Sheetlike, crystalline forms of silica obtained by extracting layer-type basic cop-
3. Flakelike gels made by freezing sols of colloidal silica or sols of hydrolyzed
I 1 obtained by the hydrolysis of calcium silicide.
per silicate with acid (92, 93).
HSiCI, and oxidizing the product (93).
These hydrated silicas are similar only i n physical form and are entirely different i n structure. (The type made by freezing is amorphous to X-rays and is considered in the following section.) A comparison of the chemical behavior of types 1 and 3 has been made by Kautsky and Reise (94).
Kautsky and associates (92, 93) have studied in some detail the lepidoidal silica obtained from a synthetic layerlike colloidal copper silicate which they formulate as [(Si60,)(0H),,][0Cu0H]. It has a very high specific surface area similar to the cop- per mineral chrysocolla (95, 96), which has a specific surface area greater than 300 m2 g- l . The corresponding silica has ion-exchange properties, but all the original copper cannot be put back into the structure once i t has been removed.
Amorphous Silicas
Amorphous silica may be broadly divided into three types:
I . Vitreous silica, made by fusing quartz. 2. Silica M , an amorphous silica formed when either amorphous or crystalline
silicas are irradiated with high speed neutrons. The density of amorphous silica

22 The Occurrence, Dissolution, and Deposition of Silica
increases and that of crystalline silica decreases. I t is thermally unstable and is transformed to quartz at 930°C i n 16 hr. Actually the silica M from different crystalline forms may differ slightly from each other. The density is 2.26, a s com- pared with 2.20 for vitreous or microamorphous silicas (53).
3. Microamorphous silica, which includes sols, gels, powders, and porous glasses which generally consist of ultimate particles less than a micron in size or have a specific surface area greater than about 3 m2 g- ’ . (These are discussed in detail in Chapters 4 and 5.)
Frondel (97) states that amorphous silica is not truly amorphous but consists of regions of local atomic order, or crystals of extremely small size, which by careful X-ray diffraction studies appear to have the cristobalite structure. Nevertheless, by ordinar) diffraction procedures this material gives only a broad band, with no multiple peaks a s are ordinarily obtained with macroscopic crystals, and is referred to here a s “amorphous.”
In nature. microamorphous silicas have either been condensed from the vapor phase ejected in volcanic eruptions or deposited from supersaturated solutions in natural waters and in living organisms. Except for silica deposited in plants or diatoms. natural microamorphous silicas are generally too impure to permit a study of solubility. (The formation and properties of natural opal are discussed in Chapter 4.)
Microamorphous silicas made in the laboratory can be divided into three classes:
1 , Microscopic sheets, ribbons, and fiberlike forms obtained by special processes. 2 . Common amorphous forms consisting of ultimate spherical particles of SiO, less
than 1000 A in diameter, the surface of which consists of anhydrous SiO, or SiOH groups. These particles may be separate or linked together in three-dimen- sional networks as shown in Figure I .2: ( a ) Discrete or separate particles as in sols. (b) Coherent three-dimensional aggregates with siloxane bonding at the points of
(c) Voluminous three-dimensional open networks of aggregated particles as in
3. Hydrated amorphous silica, here designated a s a separate class in which most, if not all, of the silicon atoms each retains one or more hydroxyl groups in the silica structure. This type o f polymeric structure is obtained i f monosilicic acid or oligosilicic acids in water are concentrated and polymerized a t ordinary or low temperature and in slightly acidic solution. I t is now believed that under these conditions the silica polymerizes to extremely small spherical units less than 20-30 A in diameter, which. when concentrated, link together into a three-dimen- sional gel mass, trapping water i n the interstices, which are of molecular dirnen- sions and retain water which can be desorbed only above about 60°C.
contact, a s i n gels.
aerogels, pyrogenic silicas, and certain dispersible silica powders.
Ordinarily such structures are not retained because i n making sols and gels the pH is

Phases of Silica
A
23
C
Figure 1.2. three dimensions but here represented only in two. A , sol; E , gel; C, powder.
Ultimate particles in common forms of colloidal silica. Aggregation is actually in
not kept sufficiently acidic nor is the temperature kept below 60°C during process- ing to the final state.
Microscopic Sheef. Ribbon, and Fiberlike Forms
Sheet or flakelike amorphous silica particles are obtained in various ways:
Formation a t the gas-liquid interface: hydrolysis of gaseous SiF, and simultaneous polymerization of the silicic acid i n water a t 100 or 0°C produced opaque “scales” of silica; so does hydrolysis of SKI, vapor a t I00”C (98). The scales appear t o be films of silica gel formed a t the gas interface when the extremely reactive vapors meet the water surface. The remarkably “fluffy” character of the powder made from SiF, by Jacobson (99) is shown by its very low bulk density of 0.025 g cm-$ and the fact that it appears to flow as water does. The irregular gel flakes, a micron or so in diameter and perhaps a tenth as thick, contain 92.86% SiO, and 7.14% H,O. Formation by freezing silica sols: When a solution of colloidal silica or polysilicic acid is frozen, the growing ice crystals exclude the silica until it remains as a concentrated sol between the ice crystals and then polymerizes and forms a dense gel. When the ice is melted the silica is obtained usually a s irregular flakes

24 The Occurrence, Dissolution. and Deposition of Silica
formed between the flat surfaces of the ice crystals. Lepidoidal silica formed in this way from polysilicic acid of low molecular weight has been extensively studied by Kautsky and associates (92-94). The gel structure is of high density owing to the high concentration of the silicic acid before i t polymerizes to gel, and the specific surface area of the microporous mass is around 900 m2 g - l s o the ultimate silica particles can only be 20-30 A in diameter. Kautsky reported that the vacuum-dried powder contained about 10% H,O; i f the water was all present as SiOH groups and each O H group occupied 12.5 8, on the surface of the ulti- mate particles, then the specific surface area is calculated to be 930 m 2 g- ' S i 0 2 . Kautsky investigated the capacity to exchange hydrogen for copper ions from solution (0.1 M Cu(NH,),SO, + 0.3 M NH,OH) i n 200 hr. The product contained one copper atom per silicon (-SiOCuOH) and had surface area (by BET-method adsorption) of 870 m2 g - ' .
3. Leaflets of silicon oxyhydride (HSiO, 5 ) up to 5 m m i n diameter are formed when HSiCI, i n ether is hydrolyzed by very gradual addition of a theoretical amount of water. The product appears to "crystallize" when the concentrated ether solution is evaporated. I t is possible the silica forms a t the silica-water interface but remains invisible because of the close refractive index until the ether evaporates (100).
4 . Sheets o f silica consisting of a single layer of colloidal particles are formed when silica is coagulated under the influence of cationic surfactants (101). The mechanism of formation is discussed in Chapter 4 ,
5. Highly elongated forms of silica. other than fibers formed from glass, have been obtained either by conversion from fibrous precursors or by unidirectional craz- ing or cracking during drying of thin films of sols.
Silicon monoxide has long been known to oxidize to fibrous silica. Nemetschek and Hofmann (102) investigated the material condensed from vapor from the reaction of silica and silicon metal. Under the electron microscope i t was found to consist of a mat of remarkable hollow tubes and spiral fibers of amorphous silica less than 0.04 micron i n diameter and many microns long. The morphology was compared wi th the fibers of halloysite and chrysotile (103).
Silica W. an unstable crystalline silica fiber (65). as previously mentioned, is converted to amorphous silica fibers by traces o f moisture. These are very similar to those obtained via silicon monoxide.
Fibers of amorphous anhydrous siiica, 1-50 microns in diameter, grow outward from an electrically heated platinum surface exposed to nitrogen-diluted SiF, and water vapor at I I00"C. according to Haller (104). The mechanism of formation is u n k n o w n . I t may be that the silica is being deposited a t the base of the fiber simply because the temperature is highest at the platinum surface under the end of the fiber around which silica vapor is condensing. Once a nucleus of viscous silica is formed it probably does not wet the platinum, so that surface tension pulls it up into a drop, the outer, cooler portion o f which solidifies and moves away. while more silica is added at the hotter base. This suggested mechanism is consistent with the observa-

Phases of Silica 2s
tion that there are often spherical beads on the end of the fiber and that the fibers wave about as they are growing.
Common Amorphous Forms
The formation and properties of silicic acids, sols, gels, and powders are described in subsequent chapters in detail, but a brief description of the nature of microamor- phous silica should preface a discussion of solubility.
There are two broad classes, as follows.
I . Anhydrous amorphous silica particles formed a t high temperature, such as “pyrogenic” or “fumed” silicas, are recovered from the gas phase as voluminous, extremely finely divided powders. They are made in one of the following ways: (a) Vaporizing silicon dioxide in an arc or plasma jet and condensing it in a
(b) Oxidizing the more volatile silicon monoxide in the vapor phase with air and
(c) Oxidizing silicon compounds in the vapor state, such as SiH,, SiCI,, or
When water vapor is present the surface of the particles may be partly hydrated as SiOH groups.
2. Surface-hydroxylated amorphous silica particles are nucleated and grown from aqueous solution supersaturated with monomer, Si(OH),. The unique feature of this system is that, unlike a solution of sugar from which sugar molecules crystallize unchanged, Si(OH), does not crystallize as such, but must undergo a dehydration reaction to form SiO,. This reaction is slow a t ordinary temperature, so that Si(OH), can deposit SiO, on the surface of a growing quartz crystal only from extremely dilute solution and a t an extremely slow rate. If the concentration of Si(OH), exceeds about 2 x M , condensation to polysilicic acids occurs, as previously described, and colloidal particles are formed.
stream of dry inert gas.
condensing the SiO,.
HSiCIS, with dry oxygen or in a hydrocarbon flame.
Ordway (105) has shown by molecular models that tetrahedral networks of silica built up by the condensation process from Si(OH), are amorphous and are spherical, unless a nucleus of crystalline structure is initially present. However, it is obvious that i f the rate of condensation of Si(OH), onto the crystaline nuclei cannot keep up with the rate of addition of Si(OH), to the system, the latter will accumulate until amorphous networks are nucleated.
Under almost all conditions, soluble silica comes out of solution as spherical, amorphous particles that, depending on concentration, temperature, and pH, remain as a sol, are aggregated into a gel network, or are coagulated as a precipitate.
As initially formed, the amorphous particles may contain some uncondensed SiOH groups within the amorphous SiO, network. but i n further processing these are largely eliminated, leaving only S iOH groups on the surface.

26 The Occurrence, Dissolution. and Deposition of Silica
Microamorphous silica i s not easily crystallized. When an ionic material such a s a salt is rapidl! precipitated from a highly supersaturated solution, it may initially be amorphous. but i t rapidl! rearranges to the ordered crystalline state. However. in the cast: of silica i n uhich bonds are largely covalent, such rearrangement can occur only at elevated temperature or in the presence o f a solvent such as water under hydrothermal conditions. Silica. in effect, is a polymeric material. Walton (106) has pointed out \ t h y an intermediate amorphous phase is likely to be precipitated i f the material is of high molecular weight or pol),meric
Carr and F>fe (57) observed that amorphous silica in water crystallizes via cristo- halite and keatite to quartz at 335°C in 830 hr under 15.000 psi pressure, hut in I8 hr at 15.000 psi.
H \tdrarrd A mnrphoits Silica
As uill be seen, the highl! hydrated silicas. uhich a re generally stable up to 60°C, must be considered sepdratel) because the! seem to differ in solubility from the anhldrous or on14 surface-hydrated forms
Two solid insoluble hbdrated amorphous silicas were made by Signer and Gross (107) by starting with ciclohex\l esters of the formulas
The first is a ring tetramer; the second is a cage-like sphere of silica in which each silicon atom is linked to one ester group. By removing the cyclohexyl groups with anhldrous H I . they obtained white amorphous powders which were extremely hy- groscopic. corresponding to
[(HO),SiO], and [HOSiO, ,I,
I t can be seen that even i f the low molecular weight silicic acid counterparts were initiallk produced. condensation of silanol groups between adjacent molecules, form- ing siloxane bonds and ua te r molecules, could occur rapidly. However, the water is still held so tenaciously that the overall chemical analysis is not changed. while the mass becomes cross-linked and insoluble.
Natural opal may contain from 5.25 to 13.7% water, which is locked within the structure and does not evaporate (108). Opal is generally nonporous in the sense that i t does not adsorb gases or liquids.
Highlq hydrated silica gels which retain water tenaciously at ordinary temperature can be obtained by reacting hydrated sodium silicate crystals with anhydrous acid so that the silicic acid is liberated and simultaneousl) polymerized in a very dense state ( 109).
Similar gels were obtained b j hydrolyzing ethyl silicate i n distilled water a t 17°C to obtain a sol containing about 0.5% SiO,. After a few days this became viscous as aggregates *ere formed. which then formed a precipitate that was filtered off as a wet gel and stored over ua t e r at 15-17°C. Removal of all traces of HCI from this

Phases of Silica 27
ester retards hydrolysis and is not even desirable. Unfortunately, the pH was not recorded but was probably below 6 if traces of acid were present ( 1 IO, 1 I I ) . When the wet gel was dehydrated under vacuum at II'C, a plot of residual water versus vapor pressure in the range from 7 to I torr revealed definite steps corresponding to 2.5, 2.0, 1.5, 1.0, and 0.5 molar ratios of water to silica.
From the vapor pressure of water at each stage of hydration, corresponding to 0.5, 1.0, 1.5, and 2.0 molar ratios, measured a t a series of temperatures from 35 to 6OoC, Thiessen and Koerner found a linear relationship between log p and T-I, and calculated the average heat of hydration, h, per mole of water over the temperature range T , - T,:
where R = gas constant P k , , Pkl = dissociation pressure of the hydrate at TI, T, P,,, P,, = vapor pressure of water a t T,, T,
h was 1.2-1.5 kcal mole-' H,O. It was mentioned earlier that Greenberg (13) found for the reaction
SiO,(amorphous) + 0 7 H,O(I) = H,SiO,(aq) AH kcal mole-' = 2.65 * 0.28
If one assumes the heat of solution of H,SiO, to be negligible and that M = 2, then A H per mole of water would be 1.32 f 0.14, which agrees with the above value obtained by quite a different method by Thiessen and Koerner.
The formation of definite hydrates in a system now known to be amorphous is dif- ficult to accept. The stepwise removal of monolayers of adsorbed water from a large fixed area of silica surface is more plausible if it can be assumed that the removal of one layer, by coincidence, corresponds to about 0.5 mole of water per silicon atom. I f this is so, no formation of water by condensation of SiOH groups is involved, and the above relation to Greenberg's data is only a coincidence.
Based on what is now known of this system, as is discussed in Chapter 3, it is likely that the sol made by hydrolyzing ethyl silicate contains polysilicic acid or silica particles that are so small that a substantial fraction of the silicon atoms are a t the surface of the particles and bear O H groups.
Initially, such particles aggregate into short chains to form a very open three- dimensional network of gel filling the aqueous phase. As water is removed the struc- ture gradually collapses, but at ordinary temperature there is a layer of water probably several molecules deep associated by hydrogen bonding to the silanol sur- face. Thus a hydrated silica gel is obtained from which the vapor pressure of water is less than that of liquid water. The stepwise removal of water may correspond to the removal of successive layers of water molecules from the silica surface. As this water is removed from between the silica particles the gel structure shrinks and the parti-
28 The Occurrence, Dissolution, and Deposition of Silica
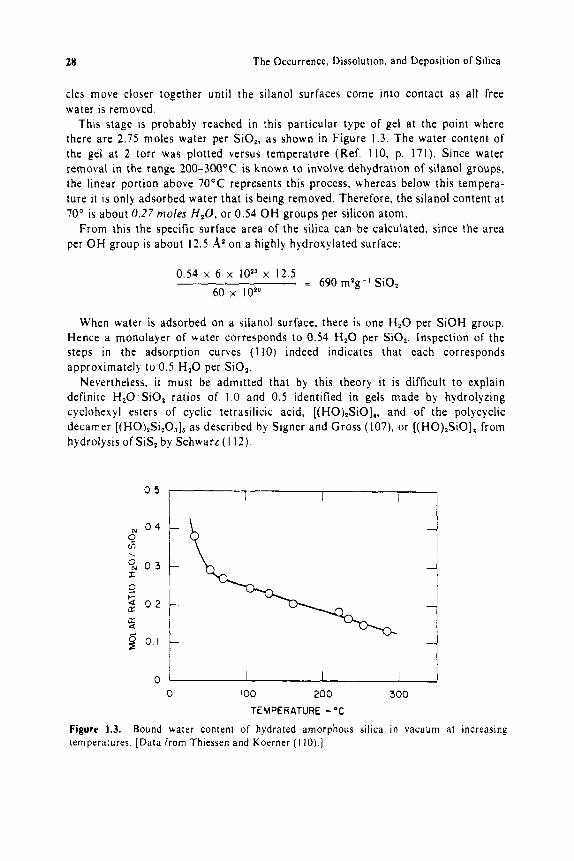
cles move closer together until the silanol surfaces come into contact as all free water is removed.
This stage is probably reached in this particular type of gel at the point where there are 2 . 7 5 moles water per SiO,, a s shown i n Figure 1.3. The water content of the gel at 2 torr was plotted versus temperature (Ref . I IO, p. 171). Since water removal in the range 200-30O0C is known to involve dehydration of silanol groups, the linear portion above 7 0 ° C represents this process, whereas below this tempera- ture it is only adsorbed water that is being removed. Therefore, the silanol content a t 70" is about 0.27 moles H,O, or 0.54 O H groups per silicon atom.
From this the specific surface area of the silica can be calculated, since the area per O H group is about 1 2 . 5 A' on a highly hydroxylated surface:
0.54 x 6 x IOz3 x 12 .5 60 x IOza
= 690 rnlg-' SiO,
When water is adsorbed on a silanol surface, there is one H,O per SiOH group. Hence a monolayer of water corresponds to 0.54 H,O per SO,. Inspection of the steps in the adsorption curves ( 1 IO) indeed indicates that each corresponds approximately to 0 . 5 H,O per SO,.
Nevertheless, i t must be admitted that by this theory it is difficult to explain definite H,O:SiO, ratios of 1.0 and 0.5 identified in gels made by hydrolyzing cyclohexyl esters of cyclic tetrasilicic acid, [( HO),SiO],. and of the polycyclic decamer [(HO),Si,O,]s as described by Signer and Gross (107), or [(HO),SiO], from hydrollsis of SiS, by Schwarz ( I 12) .
05 I I I
N O 4 1 0 cn . I 0 , 0 3 1 I
- b
I 1
I I I 0 100 200 300
TEMPERATURE -"C Figure 1.3. Bound water content of hydrated amorphous silica in vacuum at increasing temperatures [Data from Thiessen and Koerner ( I I O ) . ]

Phases of Silica 29
On the other hand, removal of layers of water from an aluminum oxide surface i n dis- tinct steps has been noted by de Boer ( I 13a). Thus the foregoing explanation cannot be ruled out. since highly hydrated gels made by neutral or low pH and at room temperature generally have specific surface areas i n the range 600-800 m2 g - ’ ,
Biogenic Silicas
These are of many types found i n widely different kinds of living organisms as isolated particles, skeletal structures, and surface elements. I n most cases, when freed from organic matter, the silica exhibits characteristic patterns and shapes (see Chapter 7) . Essentially all biogenic silica is amorphous. I t often has a substructure of extremely small particles less than 50 8, in diameter which have a surface of S iOH groups. These are joined together in close-packed three-dimensional struc- tures, some of which are isolated microscopic masses; others are solids permeated by holes like Swiss cheese; and still others are like an interconnected mass of rods. The ultimate particles i n this size range can coalesce into denser structures as the intervening pores become finer. Further deposition of silica can obliterate the particulate appearance and lead to an impervious solid. Thus specific surface areas observed in biogenic silicas vary widely from several hundred m2 g-I to very low values i n those cases where the porosity has collapsed until the pores no longer admit the nitrogen used for the measurement. These wide variations in structure and porosity are also seen in synthetic silica gels but the latter do not exhibit the charac- teristic patterns usually seen in biogenic silicas.
The biogenic silica structure must be interpenetrated by organic tissue to effect the deposition. Also, where the silica plays the role of a stiffening and strengthening agent it is interlaminated or interpenetrated by biopolymer: in plants by cellulose, or i n diatoms by a protein, silicalemma.
Many microcrystalline silica minerals such as flint, chert, and chalcedony can be formed from biogenic silica by compaction and microcrystallization. Kieselguhr (diatomite), which was originally amorphous, is found in various stages of transformation. The changes over millions of years can be noted, for example, by the decrease in solubility and changes in other properties. Furthermore silica undergoes continuous dissolution and biogenic redeposition i n the oceans. Hurd and associates ( I 13b-e) studied the properties of biogenic silica of ages up to 40 million years, with emphasis on differences in solubility, density, and refractive index, com- paring these with the values for known crystalline forms. which were thoroughly reviewed.
Kastner, Keene, and Gieskes ( 1 130 have examined the transformation of amorphous biogenic silicas in the form of siliceous “oozes” on the ocean bottom to partially crystalline geological deposits. They reviewed the complex background literature and gave experimental data showing more rapid transformations when the silica was embedded in carbonate (chalk). The remarkable 80 million year shrinkage of the silica skeletons of large sponges to dense round boulders of flint in the chalk beds of England is probably an example of this phenomenon.

30 The Occurrence. Dissolution, and Deposition of Silica
T H E SOLUBILITY OF SILICA
Before the solubility of microamorphous silica, which is a basic factor in the behavior of silica as a colloid, is considered in detail, the solubility of crystalline phases of silica is briefly reviewed.
Solubility of Quartz at Ordinary Temperature
Prior to the outstanding investigation by Van Lier, published in 1960 ( 1 14, 115), there was speculation that quartz might exhibit no true equilibrium solubility in water at ordinary temperature, although Gardner ( I 16) reported 6 ppm in 1938.
Solubility data were very confusing until i t was found that traces of certain metal impurities and especially the presence of an amorphous or at least disturbed layer on the crystal surface caused variable results. especially a t temperatures below 150°C. In 1952, Dempster and Ritchie (117) reported that siliceous dusts have a layer of high solubility that gradually blends into the solid core, which adsorbs basic dye- stuffs ( I 18). Alexanian ( I 19) found by electron diffraction that quartz possesses a surface layer of amorphous silica about 100 A thick, which is removed by H F but is re-formed in ambient humidity. Waddams found that the quartz surface i n water released “mosaic” silica, presumably as particles of colloidal size, since they scat- tered light (120). This was confirmed by Sakabe et al . ( IZI) , who found that in neu- tral or alkaline aqueous suspension, quartz released both soluble silica and colloidal particles of crystalline nature, 0.01-0.3 microns in size. Stober and Arnold (122) found that the amount of silica released was much more than a monomolecular layer. and that i t decreased with successive changes of water. When quartz was intensively pulverized in water, the disturbed surface layer can amount to a s high a s 35%. with a specific surface area of 70 m z g-l , and the solubility is increased from less than I O to 70 ppm at 25°C (123). Paterson and Wheatley (124) made similar observations.
The disturbed layer on ground quartz particles 1.5 microns in diameter was examined by Koopmans and Rieck (125). by gradually dissolving it in dilute H F while following the X-ray peak intensity. The layer thickness was 0.1-0.2 microns. Similar studies by Lidstrom (126). who also used nmr, indicated a disturbed layer up to 2 microns thick may exist, but upon aging in water and removal of the outer por- tion, the remaining underlying disturbed and strained layer returned to the normal crystalline state.
Cleaning the Surface
Van Lier ( 1 14) studied the dissolution of ground quartz of particle size 3-15 mi- crons. He found a more soluble disturbed surface layer, 0.3 microns thick, which could be removed by stirring IO g of the powder into 50 ml 9-15% H F for 5 min to dissolve 25% of the silica; then the residue was washed with 0.1 N N a O H and then water to remove fluoride and alkali, and dried and stored in a desicator. When this powder was suspended i n 0.1 N N a O H a t 26”C, the rate of dissolution remained
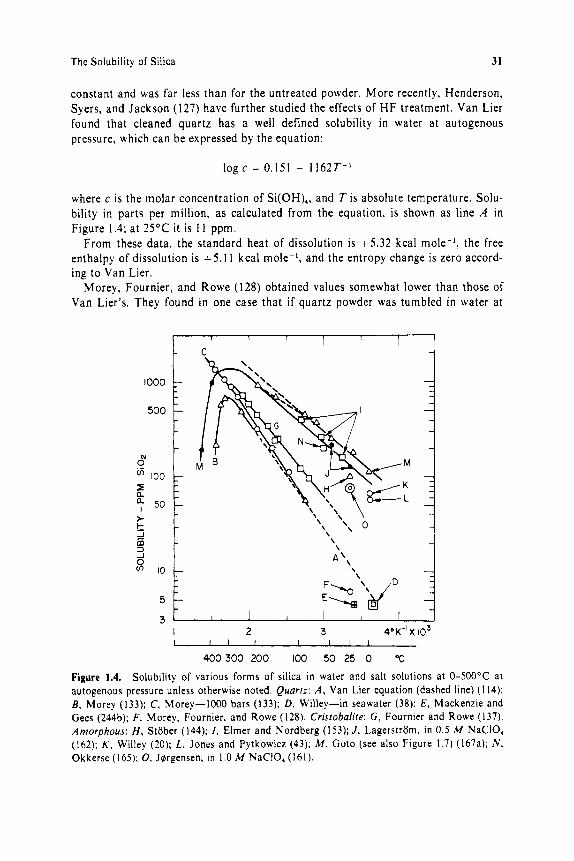
The Solubility of Silica 31
constant and was far less than for the untreated powder. More recently, Henderson, Syers, and Jackson (127) have further studied the effects of H F treatment. Van Lier found that cleaned quartz has a well defined solubility in water a t autogenous pressure, which can be expressed by the equation:
log c = 0.151 - 1 l 6 2 T - ’
where c is the molar concentration of Si(OH),, and T i s absolute temperature. Solu- bility in parts per million, as calculated from the equation, is shown as line A in Figure 1.4; a t 25°C it is I 1 ppm.
From these data , the standard heat of dissolution is + 5 . 3 2 kcal mole-’, the free enthalpy of dissolution is + 5 . 1 I kcal mole-’, and the entropy change is zero accord- ing to Van Lier.
Morey, Fournier, and Rowe ( 1 28) obtained values somewhat lower than those of Van Lier’s. They found in one case that i f quartz powder was tumbled i n water a t
1000
500
0” Gi 100 z a 4- 50
m
* IO
3 0
5
3
t \ \
A‘\
I 2 3 4’K-lX IO3 1 1 1 I I I l l
400 300 200 100 50 25 0 T Figure 1.4. Solubility of various forms of silica in water and salt solutions at 0-500°C at autogenous pressure unless otherwise noted. Quarrz: A , Van Lier equation (dashed line) ( I 14); B , Morey (133); C, Morey-I000 bars (133); D , Willey-in seawater (38): E , Mackenzie and Gees (244b); F, Morey. Fournier. and Rowe (128). Cristobalite: G, Fournier and Rowe (137). Amorphous: H, Stober (144); I , Elmer and Nordberg ( I 53); J , Lagerstram. in 0.5 M NaCIO, (162): K , Willey (20); L, Jones and Pytkowicz (43); M , Goto (see also Figure 1.7) (167a); N . Okkerse (165): 0, Jergensen, in 1.0 M NaCIO, (161).

32 The Occurrence, Dissolution. and Deposition of Silica
25"C, the silica increased to 80 ppm after a year, and so was highly supersaturated, and then dropped to 6 ppm, the true solubility. It is not known why the quartz sur- face already present could not accept the silica, but new nuclei had to be created to reach eq u i I i br i u m .
Siever (129) also investigated the solubility of quartz with results compatible with those of Van Lier.
Solubility of Quartz Under Hydrothermal Conditions
I t is impossible to review the extensive investigations i n this highly specialized field. Some of the publications are a s follows.
Kennedy and associates developed data against which subsequent work was often compared (25, 130, 131). In 1954, extensive studies were made by Wyart and Sabatier ( l32) , who measured the solubility of quartz, tridymite, cristobalite, and vitreous silica in both water and steam phases a t up to 480 bars and 470°C. Increased solubility of quartz with pressure had earlier been examined up to 600°C by Morey (133). and more recently by Heitmann (30); Anderson and Burnham (134) examined solubility i n water and salt and alkali solutions up to 900°C and 6 kilo- bars. Solubility was only slightly reduced by the presence of salt, but increased in direct proportion to the base added. The solubility of quartz under hydrothermal conditions is described i n three papers by Kitahara (135) with special attention to supercritical conditions up to 500°C and 900 bars. The heat of solution of quartz calculated from solubility data was 7.8 kcal mole- ' . In 1965, Heitmann (30) surveyed the solubilit) of silica in water and stream and assembled data based on more than 1000 experiments up to 650°C and 300 kg cm-*, on the basis of which a complete solubilit) diagram was constructed. Verifying earlier work, the solubility of silica is shown to increase with increasing density of steam or water, and reach a maximum near the critical point of water. The thermodynamic properties and solubility of quartz up to 600°C and 5 kilobars pressure are being summarized by Hegel- son ( I 36).
Solubility of Cristobalite and Tridymite
There is some question whether true equilibrium can be established for cristobalite and tridymite. There appears to be no instance where these crystals have been grown under hydrothermal conditions in preference to quartz. Fournier and Rowe (137) believed that precipitation of crystalline silica under hydrothermal conditions would generally result i n quartz. For this reason, i n their study of the solubility of cristo- balite, they left water in contact with the solid for long times without changing temperature. so as not to supersaturate the solution any more than necessary, and then suddenly quenched the system and measured the concentration of monomeric silica, which was the only type of silica i n the water. I n Figure 1.4, line G strongly suggests that a definite solubility value was established.

The Solubility of Silica
From the slope of the line,
33
Thus
-AH0 l o g s = 4.576T - + constant
and
AH = 4.58 kcal mole-'
where A H is the differential heat of solution of cristobalite. The solubility at 25"C, by extrapolation, is 27 ppm, or 0.45 millimolal.
whence
AF;',o, = 4.56 kcal mole- '
From their corresponding data for the quartz system.
AH2@,oK = 6.0 kcal mole-l
AFfgUoK = 5.46 kcal mole-.'
Thus the free energy change going from cristobalite to quartz was estimated to be -0.90 i 0.3 kcal mole-l, which is in reasonable agreement with 0.57 i= 0.75 kcal mole- ' calculated from the heats of formation from elements:
Quartz: A F o = 196.582 * 0.30 Cristobalite: A F o = 197.151 i 0.75
Solubility of Other Crystalline Forms of Silica
For measuring the solubility of silica in water Stober (138) adopted a standard buf- fered salt solution (Ringer's solution) containing 0.9% NaCI, which expedites equili- bration but does not affect solubility, and 0.1 % NaHCO,, which buffers the solution a t pH 8.4, where dissolution occurs most rapidly without appreciable formation of silicate ions that would occur at higher pH.

34 The Occurrence, Dissolution. and Deposition of Silica
Stober compared the solubilities of different crystalline forms using ground powders of different particle sizes and treated with N a O H solutions to remove disturbed material from the crystal surfaces. Dissolution behavior proved to be very complex. Solrrbilirj. in the sense of a djlnaniic equilibriirnr between silica in solution and i t i rhe solid phase upparent1.i. e.visrs on!,, with crystalline quartz, and amorphous or r~irreoirs silica ( 1 39).
The dissolution of the other crystalline phases. crystobalite, coesite. tridymite, and stishovite, with increasing concentration of Si(OH), is accompanied by adsorption of Si(OH), on the c r y t a l surface. which inhibits fu r the r dissolution. Thus the final concentration of soluble silica depends on relative rates of dissolution and adsorp- tion, and ma) reach the limiting solubility of amorphous silica.
Stishovite. H hich differs from the other forms by having a more dense octahedral structure. with a higher surface concentration of S iOH groups. behaved most pecu- liarl!. I n 0. IC; H F solution i t was completely insoluble. coesite dissolved slowly, and quartz dissolved most rapidly. Yet in Ringer's solution quartz reached its equilib- rium solubilit? of about I2 ppm in a few dajs . but coesite continued to dissolve for a month 2s the silica concentration reached more than 25 ppm, and stishovite dissolved rapidly un t i l the soluble silica reached 190 ppm, a t which point amorphous silica was apparently nucleated and the silica concentration began to decrease (140).
Extensive further studies were summarized by Stober (141). and a theor! of solu- bilit! \+as formulated.
Adsorbed Silica on Crystalline Silica
Holt and King discovered i n 1955 that when powdered quartz was added to water, the concentration of dissolved silica, Si(OH),, increased rapidly at first but thereafter ver) s lou l j . This was traced to a form of silica that amounted to 16%' of a monolayer on the crystal surface and that dissolved rapidly. Furthermore, when this material was removed b> treatment with alkali. the cleaned surface then adsorbed soluble silica from solution. to reestablish the layer. Using %(OH), containing radioactive 31Si. the) found that a t pH 5 it did not exchange with the surface; at pH 9 i t exchanged wi th adsorbed Si(OH),. but not with the cleaned crystal surface (142. 1-13),
Stober investigated the solubility behavior of several modifcations of silica and developed a theor) and equation for the behavior of adsorbed monosilicic acid in retarding or preventing approach to true solubility equilibrium (144).
Stober believed that the interaction of Si(OH), and the silica surface involved the following steps:
I . At the surface Si-0-Si bonds are split b j hydrolysis. averaging two b o n d i n g per tetrahedra. forming a hkdrated silicic acid molecule adsorbed on the surface.
2 . The Si(OH), is desorbed into solution. 3. Si(OH), is adsorbed onto the surface at equilibrium as the reverse process occurs,
folloi+ed b \ condensation and addition of SiO, to the surface.

The Solubility of Silica 35
He formulated the following equations. For the adsorption of Si(OH), the equation is
n,bc 1 + bc
nods = -
where nods = surface concentration of adsorbed Si(OH), no = surface concentration of complete mololayer c = silicic acid concentration in solution b = adsorption constant
Adsorption occurs only on the bare surface not covered by Si(OH),:
where nH = silicic acid released by hydrolysis per unit of surface area f = time
k = kinetic constant for hydrolysis A = activation energy of hydrolysis R = gasconstant T = temperature ( O K )
It is assumed adsorption equilibrium is established rapidly relative to the other steps, Thus equation I can be inserted into equation 2:
- - - - n0 k e x p -6) dn, dt 1 + bc
Condensation involves adsorbed molecules only:
dnc - = -nadsk' exp -(&) dr
(3)
(4)
where n, = silicic acid condensed per unit of surface area k' = kinetic constant for condensation A' = activation energy for condensation
Since temperature is assumed constant and values of k , k ' . A , and A ' do not change, new constants can be introduced:

36 The Occurrence, Dissolution. and Deposition of Silica
Combining equations gives
( K - K ' b c ) dn dnH dn, no - = - + - = - dr dt dt I + bc
where n is the concentration of adsorbed monomer at time t .
solvent. V , and total surface area of solid silica, F . The concentration of silicic acid, c, can be expressed in terms of volume of
n,Fbc F V(I + bc) V
c = c , -
where c, = initial concentration of Si(OH), i n solution.
in the volume. respectively: Differentiation gives the relation of changes in concentrations on the surface and
dn - = [;+-I- n& dc
di ( 1 + bc)Z dt
Substituting for dn/dr and integrating gives
c, b In(l + bc) Ybc bnoF + V(l - bc,)Z
t = - [ K I + bc, n OF noF( l + bc,) - 31 where c, is the equilibrium solubility concentration for the two-phase system soluble silica plus amorphous silica, defined as c, = K / b K ' .
The amorphous silica phase presumably might appear i n solution a s colloidal particles or as a built-up layer at some points on the crystal surface. Stober used the experimental value of c, of 1 I O ppm (micrograms of SiO, per milliliter). For no, the value of 600 k g m - z was calculated on the basis of IO micromoles m - z (145) or 6 Si(OH), nm-,.
Experimental data were obtained by measuring, at many times over 3-4 weeks, the amount of silica dissolved into 500 ml of Ringer's solution a t pH 8.2 a t 25°C from I O rn, of silica surface. Using particles of different sizes, Stober found a rela- tibely constant rate of solution per u n i t of surface area.
For quartz, he used six different sizes of particles ranging from about 0.07 to 1.3 microns with specific surface areas of 33.6-1.7 mz g-'. From the curve of concentra- tion o f dissolved silica versus time, the constants for the above equation were determined as
b = 0.7 m l w g - ' K = 1 . 7 day- '
The final concentrations of soluble silica ranged from I I to 13 ppm i n excellent agreement with Van Lier's value of 1 I ppm ( I 15) for the solubility of quartz.
The Solubility of Silica 31
For vitreous silica, b = 0.7 ml pg-’ (as for quartz) but a much higher value for the
With b = 0.7 ml p g - l assumed, the following values were found: hydrolysis constant, K , indicated a much higher hydrolysis rate than for quartz.
K (day - I ) K’(duy - l) Modijcat ion (Hydrolysis) (Condensation)
~~ ~
Quartz 1.7 0.2 Vitreous 90 1.17 Cristobalite 4.0 - Stishovite 20 -
Coesite 0.4 Tridymite 2.4 -
When the phases other than quartz and amorphous silica are suspended in water, the final concentration of soluble silica is not a saturation concentration, but the result of a competition between silica passing into solution as Si(OH), and Si(OH), readsorbing on the surface to an ever-increasing extent, blocking further dissolution. Thus a limiting concentration is reached that depends on the area of solid surface exposed per unit volume of solution.
When the same sample of silica having an area of 1 mz is put each day into 50 ml offresh Ringer’s solution (pH 8.4) for 24 hr, the amount of soluble silica dissolving each day reaches a steady value, which is characteristic of the particular modifica- tion of silica used:
Specific Surface Limiting Silica Modification (m’g-l) Concentration (ppm)
Vitreous (glass) Stishovite Cristobalite Tridyrnite Quartz Coesite
8.8 21.6
8.5 12.0 10.4 10.6
39 11 6 4.5 2.9 0.7
If Stober’s views are correct and cristobalite, for example, has no true equilibrium solubility, then calculations based on published solubility cannot be valid. The need for a more accurate definition of solubility has been pointed out by Weill and Bot- tinga (146). Data presented as “solubility” of different forms of silica is often only an indication of the rate of solution or the limiting concentration reached under particular conditions. Thus a report that opal is 15-18 times as soluble as anhydrous silica a t pH 12.8 merely indicates the relative rate of dissolution (147).
Similarly, solubility data by Kopeikin and Mikhailov (148) for quartz (7 ppm), cristobalite ( I 2 ppm), tridymite (16 ppm), vitreous silica (88 ppm), and amorphous silica (120 ppm) are, in the cases of tridymite and cristobalite, higher than the limit- ing concentrations observed by Stober, no doubt because different techniques were
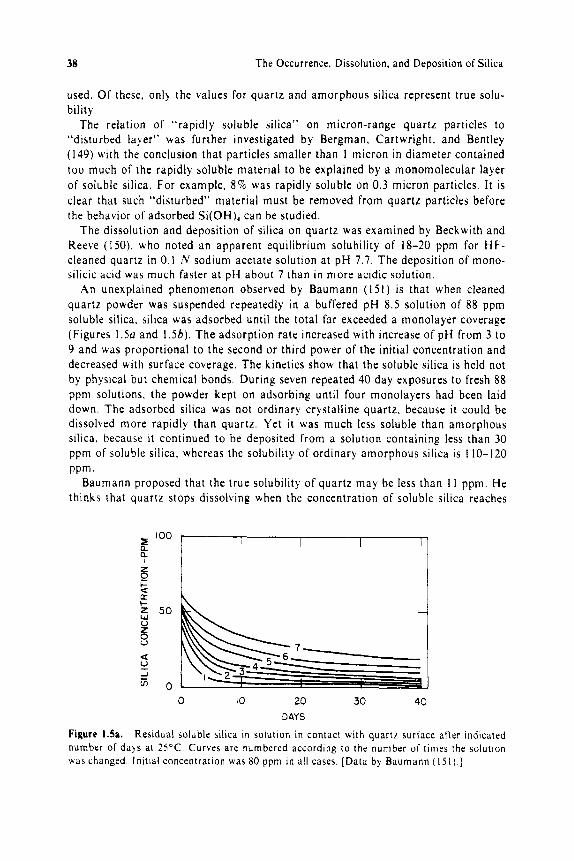
38 The Occurrence. Dissolution, and Deposition of Silica
used. Of these. only the values for quartz and amorphous silica represent true solu- bility.
The relation of "rapidly soluble silica" on micron-range quartz particles to "disturbed layer" was further investigated by Bergman, Cartwright, and Bentley (149) w i t h the conclusion that particles smaller than 1 micron in diameter contained too much of the rapidly soluble material to be explained by a monomolecular layer of soluble silica. For example, 8% was rapidly soluble on 0.3 micron particles. i t is clear that such "disturbed" material must be removed from quartz particles before the behavior of adsorbed Si(OH), can be studied.
The dissolution and deposition of silica on quartz was examined by Beckwith and Reeve (150). h h o noted an apparent equilibrium solubility of 18-20 ppm for HF- cleaned quartz in 0.1 N sodium acetate solution at pH 7 . 7 . The deposition o f mono- silicic acid was much faster a t pH about 7 than i n more acidic solution.
An unexplained phenomenon observed by Baumann (151) is that when cleaned quartz powder was suspended repeatedly i n a buffered pH 8.5 solution of 88 ppm soluble silica. silica was adsorbed until the total far exceeded a monolayer coverage (Figures 1 . 5 ~ and 1.56). The adsorption rate increased with increase of pH from 3 to 9 and was proportional to the second or third power of the initial concentration and decreased with surface coverage. The kinetics show that the soluble silica is held not by physical but chemical bonds. During seven repeated 40 day exposures to fresh 88 ppm solutions, the powder kept on adsorbing until four monolayers had been laid down. The adsorbed silica was not ordinary crystalline quartz, because i t could be dissolved more rapidly than quartz. Yet it was much less soluble than amorphous silica, because it continued to be deposited from a solution containing less than 30 ppm of soluble silica. whereas the solubility of ordinary amorphous silica is 110-120 PPm,
Baumann proposed that the true solubility of quartz may be less than 1 I ppm. He thinks that quartz stops dissolving when the concentration of soluble silica reaches
0 IO 20 30 40 DAYS
Figure 1 . 5 ~ . Residual soluble silica in solution in contact wi th quart1 surface after indicated number of dais at 25°C. Curves are numbered according to the n u m b e r o f t imes the solution was changed. in i t ia l concentration was 80 ppm i n all cases. (Data by Baumann ( I S I ) . ]
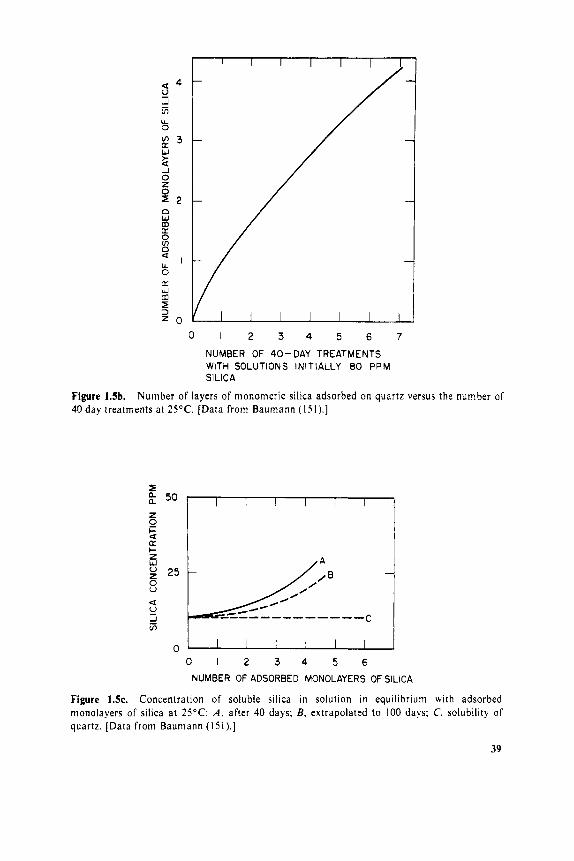
0 1 2 3 4 5 6 7
NUMBER OF 40-DAY TREATMENTS WITH SOLUTIONS INITIALLY 80 PPM SILICA
Figure 1.5b. 40 day treatments a t 25°C. [Data from Baumann ( 1 5 1 ) . ]
Number of layers of monomeric silica adsorbed on quartz versus the number of
5
50 , E
0 0 1 2 3 4 5 6
NUMBER OF ADSORBED MONOLAYERS OF SILICA
Figure 1.5~. Concentration o f soluble silica in solution in equilibrium with adsorbed monolayers of silica a t 25°C: A . after 40 days: 8, extrapolated to 100 days; C, solubility of quartz. [Data from Baumann (ISI) . ]
39

40 The Occurrence, Dissolution, and Deposition of Silica
I I ppm because at this concentration enough Si(OH), is adsorbed to stop further dissolution at 25°C. On the other hand, 1 1 ppm is the solubility value extrapolated from solubility data a t higher temperatures where i t is known that true solubility equilibrium is reached and that quartz crystals grow when the solution is supersatu- rated.
It is suggested that a t 25°C the adsorbed layers of silica are very slow to assume the ordered structure of the underlying crystal lattice, but nevertheless crystallization is s l o ~ l y occurring just as i t does more rapidly a t higher temperatures.
A possible explanation for the above observations is a s follows: the first adsorbed layer o f Si(OH), is held first by hydrogen bonds and then condensation by siloxane bonds in a fairly regular arrangement almost matching the very regular array of underlying silicon atoms. I t is the regularity of the silanol groups on the quartz sur- face that permits almost perfect molecular matching and very strong adsorption of the Si(OH),. Each subsequent layer becomes less regular, and thus the adsorption constant is lower and the apparent solubility of that layer becomes higher. Even in the fourth layer, the regularity is still so pronounced that the solubility is still only about 25 ppm.
From a plot of Baumann's data for the concentration of silica in solution after 40 days versus the number of molecular layers of SiO, (Figure 1 . 5 ~ ) it appears that after the second layer, the solubility increases with the number of layers a s the atomic arrangement becomes more disordered.
From a plot of the data i n Figure 1 . 5 ~ as parts per million in solution versus the logarithm of time (days), the more nearly linear curves permit a reasonable extrapolation to 100 days. As shown in Figure 1 . 5 ~ the amount of silica i n solution continues to drop. suggesting that in all cases i t is approaching the solubility of quartz. I t is therefore likely that i f this quartz powder with multilayers of adsorbed silica on the surface were left to age for a year or so instead o f 40 days, or i f the suspension were heated to IOO'C. for example, the adsorbed material would become further converted to the structure of quartz and its solubility decrease to that of quartz.
Solubility of Amorphous Silica
.A given sample of amorphous silica exhibits a reproducible equilibrium solubility in water. However, reported solubility values for amorphous silicas range from 70 to more than 150 ppm a t 25°C. Such variations are apparently due to differences in particle size, state of internal hydration, and the presence of traces of impurities i n the silica or absorbed on its surface during the measurements.
As a basis of comparison. it is of interest to note the solubility data for silica of which the particles are large enough that size has no effect.
Based on data for smaller particles. an extrapolation of Alexander's (152) data indicates the solubility of massive amorphous silica to be 90 ppm for silica from pure sodium silicate. and 60-70 ppm for silica from commercial water glass. which probably contains traces of impurities that reduce solubility. Similar extrapolated values are described below (Figure I . l o b ) .

The Solubility of Silica 41
Vitreous silica has the same solubility as other amorphous silica. Because of the small specific surface area of powdered silica glass i n comparison with that of microamorphous or colloidal silicas, workers found it difficult to establish solubility equilibrium. Stober (144) found that at pH 8.4 in Ringer's solution (0.9% NaCI, 0.1 90 NaHCO,) at 2 5 ° C at least 15 days was required to reach equilibrium when 20 m* of silica surface was exposed per liter, regardless of particle size. Without the use of this solution, which has an optimum catalytic effect, it would probably have been impossible to establish equilibrium. The solubility was found to be about 100 ppm.
A highly porous, vitreous silica which equilibrated much more rapidly was used by Elmer and Nordberg (153) in a study of solubility in nitric acid solutions. Their values for high dilution at pH 3 were 160 ppm at 36"C, 260 ppm at 65"C, and 400 ppm at 95°C. They found the solubility was identical to that of a commercial dried silica gel, provided care was taken not to abrade the gel by disturbing i t during the test.
There have been several surveys of the solubility of amorphous silica, as follows. In 1955, Baumann (154) concluded that amorphous silica had a uniform solubility
in the neutral pH region, that the dissolved species was almost entirely monomeric, and that i n the pH range from 3 to 6 the rate of dissolution increased linearly with increasing pH. In the same year, Krauskopf (155) presented an excellent summary of previous studies of the dissolution and precipitation of silica at moderate tempera- tures, and emphasized the recognition of the differences between soluble, ionic, and colloidal silica, relating these to the solubility behavior of silica in natural waters.
Establishment of Solubility Equilibrium
Equilibrium is established only very slowly, unless the amorphous silica is so finely divided or microporous as to furnish an area of hundreds or thousands of square meters of surface per liter of water. Stober (144) has shown that Si(OH), is adsorbed on the surface of amorphous silica, retarding its dissolution, as has already been described in the case of quartz. The surface of vitreous silica appears to consist of SiOH groups, and if !%(OH), is absorbed by hydrogen bonding and then is partially condensed the surface may then at least partly consist of =Si(OH)* groups, referred to as vicinal hydroxyl groups. Depending then on the state of the surface of amor- phous silica before it is isolated and dried, there may be found different numbers of hydroxyl groups per unit of surface area.
Likewise, the amount of silica passing into solution when a powder is first placed in water may depend on its state before it was dried. However. this should not affect the final equilibrium solubility of the silica.
According to Baumann (l54), when amorphous silica powder (Aerosil) is placed in water at 25"C, the approach to equilibrium is different at high and low pH. Below pH 7. the concentration of soluble silica increases for several days and approaches the final solubility value asymptotically. Above pH 7, the silica concentration rises rapidly in the first day to form a supersaturated solution containing, for instance, 155 ppm at pH 9. Then the concentration drops over a period of 3-4 days to the solubility characteristic of the pH and type of silica, such as 125 ppm.
Baumann proposes that the initial rate of dissolution at this pH is greater than the
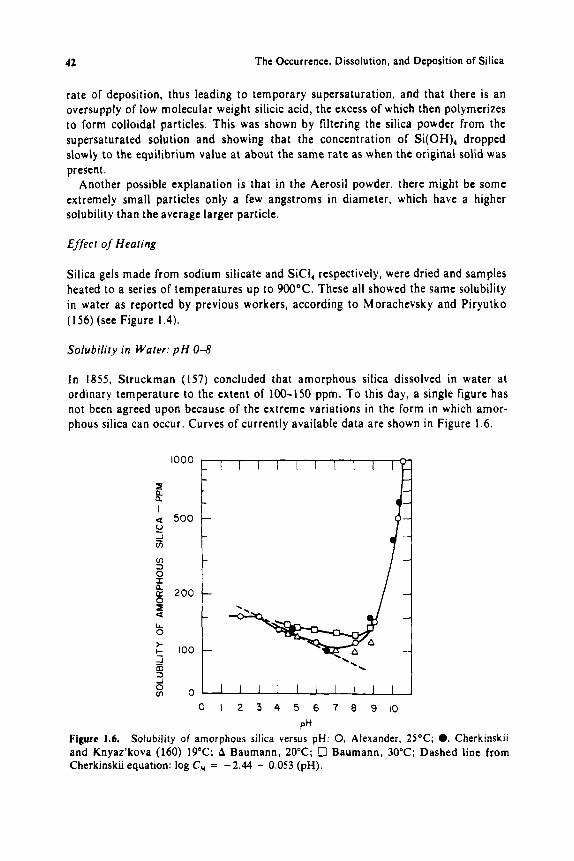
42 The Occurrence, Dissolution, and Deposition of Silica
rate of deposition, thus leading to temporary supersaturation, and that there is an oversupply of low molecular weight silicic acid, the excess of which then polymerizes to form colloidal particles. This was shown by filtering the silica powder from the supersaturated solution and showing that the concentration of Si(OH), dropped slowly to the equilibrium value at about the same rate as when the original solid was present.
Another possible explanation is that in the Aerosil powder, there might be some extremely small particles only a few angstroms i n diameter, which have a higher solubility than the average larger particle.
EfJect of Heating
Silica gels made from sodium silicate and SiCI, respectively, were dried and samples heated to a series of temperatures up to 900°C. These all showed the same solubility in water as reported by previous workers, according to Morachevsky and Piryutko ( 1 56) (see Figure I .4).
Solubility in Water: p H 0-8
In 1855, Struckman (157) concluded that amorphous silica dissolved in water at ordinary temperature to the extent of 100-150 ppm. T o this day, a single figure has not been agreed upon because of the extreme variations in the form in which arnor- phous silica can occur. Curves of currently available data are shown i n Figure I .6.
1000
L? n I Q 500 9 -J m v) 3 0 I
0 200
5 LL 0 t c_ 100
Li 3 $ 0
1 1 1 1 I l i i t i
I
0 1 2 3 4 5 6 7 8 9 1 0
PH Figure 1.6. Solubility of amorphous silica versus pH: 0, Alexander, 25°C; 0. Cherkinskii and Knyaz’kova (160) 19°C; A Baumann, 20°C; 0 Baumann, 30°C; Dashed line from Cherkinskii equation: log C , = -2.44 - 0.053 (pH).

The Solubility of Silica 43
Alexander, Heston, and Iler ( 1 5 8 ) were the first to show that microamorphous silica as powder made by collecting the smoke from burning SiH, or as colloidal silica in sol form both exhibited about the same solubility. In retrospect, these silicas probably had specific surface areas of 240 and 500 m z g - l , respectively, and the solu- bility a t pH 7-8 was about 100 and 130 ppm, respectively. A silica gel, made under conditions that we now know give a high specific surface area of more than 600 m2 g- l , was soluble to the extent of 200 ppm a t 25”C, and the dissolved silica was proved to be monomeric by chemical and physical methods.
Goto, Okura and Kayarna (159) in 1953 showed that earlier data indicating a minimum solubility a t pH 3 were in error, and that solubility was constant from pH 2 to 8, then increased rapidly a t higher pH.
Possible Solubility Minimrrm at p H 7
Baumann (154) collected data from other sources for comparison with the data of Alexander plotted in Figure 1.6; these data seem to confirm that the solubility reaches a minimum a t pH 7-8, but the reason for the slightly higher solubility at lower pH is not known. However, Cherkinskii and Knyaz’kova (160) verified the data (see Figure 1.6) and proposed that silica is amphoteric and is cationic below pH 7. They give equations assuming that all the “soluble” silica is actually polymeric and cationic a t low pH. There is no experimental basis for such a theory; all of the soluble silica has been shown to be monomeric, and there is no evidence for cationic silica above the isoelectric point of pH 2.
Solubility in Nitric Acid
Elmer and Nordberg (153) have furnished data on solubility of porous vitreous silica in nitric acid solutions up to 9 N:
log C = -0.168N - 0.332 log C = -0.167N - 0.584 log C = -0.184N - 0.796
a t 95°C a t 65°C a t 36°C
where C = concentration of SiO, (mg ml-l) N = nitric acid normality
Thus in 7 N nitric acid a t 95°C the solubility was only about 30 ppm, a s compared to 400 ppm in water, whereas a t 36OC it was 8 versus 160 ppm. I t was pointed out, therefore, that to extract impurities from amorphous silica with nitric acid, strong acid should be used. The solubilities in water ( N = 0) are included in Figure 1.4 at I .
Solubility in NaCIO, Solutions
A careful study by J ~ r g e n s e n (161) showed that there are still mysteries to be explained regarding the solubility of amorphous silica. In spite of all previous work indicating that the solubility in water is 100-120 ppm, and that solubility is not
Next Page