Embed Size (px)
DESCRIPTION
hop
Citation preview

Acta Biomaterialia 7 (2011) 3370–3381
Contents lists available at ScienceDirect
Acta Biomaterialia
journal homepage: www.elsevier .com/locate /ac tabiomat
Biocompatibility of modified polyethersulfone membranes by blending anamphiphilic triblock co-polymer of poly(vinyl pyrrolidone)–b-poly(methylmethacrylate)–b-poly(vinyl pyrrolidone)
Fen Ran a,b,c, Shengqiang Nie a,b, Weifeng Zhao a,b, Jie Li c, Baihai Su a, Shudong Sun a, Changsheng Zhao a,b,⇑a College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, People’s Republic of Chinab National Engineering Research Center for Biomaterials, Sichuan University, Chengdu 610064, People’s Republic of Chinac State Key Laboratory of Gansu Advanced Non-Ferrous Metal Materials, Lanzhou University of Technology, Lanzhou 730050, People’s Republic of China
a r t i c l e i n f o a b s t r a c t
Article history:Received 20 January 2011Received in revised form 7 May 2011Accepted 20 May 2011Available online 27 May 2011
Keywords:Polyethersulfone membraneAmphiphilic triblock co-polymerUltrafiltrationBlood compatibilityCytocompatibility
1742-7061/$ - see front matter � 2011 Acta Materialdoi:10.1016/j.actbio.2011.05.026
⇑ Corresponding author at: College of Polymer ScienLaboratory of Polymer Materials Engineering, SichuanPeople’s Republic of China. Tel.: +86 28 85400453; fa
E-mail addresses: [email protected], zhaoch
An amphiphilic triblock co-polymer of poly(vinyl pyrrolidone)–b-poly(methyl methacrylate)–b-poly-(vinyl pyrrolidone) (PVP-b-PMMA-b-PVP) was synthesized via reversible addition–fragmentation chaintransfer (RAFT) polymerization. The block co-polymer can be directly blended with polyethersulfone(PES) using dimethylacetamide (DMAC) as the solvent to prepare flat sheet and hollow fiber membranesusing a liquid–liquid phase separation technique. The PVP block formed a brush on the surface of theblended membrane, while the PMMA block mingled with the PES macromolecules, which endowedthe membrane with permanent hydrophilicity. After adding the as-prepared block co-polymer the mod-ified membranes showed lower protein (bovine serum albumin) adsorption, suppressed platelet adhe-sion, and a prolonged blood coagulation time, and thereby the blood compatibility was improved.Furthermore, the modified PES membranes showed good cytocompatibility, ultrafiltration and proteinanti-fouling properties. These results suggest that surface modification of PES membranes by blendingwith the amphiphilic triblock co-polymer PVP-b-PMMA-b-PVP allows practical application of thesemembranes with good biocompatibility in the field of blood purification, such as hemodialysis and bio-artificial liver support.
� 2011 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
1. Introduction
The aim of this study was to find an approach by which to mod-ify polymeric hollow fiber membranes with excellent biocompati-bility, which would provide information for their practicalapplication. Thus an amphiphilic triblock co-polymer was synthe-sized and directly blended with polyethersulfone (PES) to prepareflat sheet and hollow fiber membranes.
It is well known that polymeric materials are widely used in thefield of blood purification for artificial organs, medical devices anddisposable clinical instruments, including hemodialysis, hemodia-filtration, hemofiltration, plasmapheresis, and plasma collection[1,2]. Among the polymeric materials used in biomedical fields,PES is one of the most important and widely used. PES and PES-basedmembranes show outstanding oxidative, thermal and hydrolyticstability, as well as good mechanical and film-forming properties
ia Inc. Published by Elsevier Ltd. A
ce and Engineering, State KeyUniversity, Chengdu 610065,x: +86 28 [email protected] (C. Zhao).
[3]. The membranes also showed high permeability for lowmolecular weight proteins when used as hemodialysis membranes[4]. However, the blood compatibility of PES membranes is inade-quate, and injections of anticoagulants are needed during hemodial-ysis [5].
To improve the blood compatibility of biomedical materialsmany studies have focused on the development of new materialsand the modification of conventional materials. The modificationapproaches mostly used for PES membranes include blending,coating, surface physical treatment, surface grafting, etc. [6–11].Grafting anti-thrombogenic materials and/or hydrophilic polymersonto membrane material surfaces using chemical modification isthe most efficient method, which can catalytically increase the for-mation rate of antithrombin III and inhibit thrombin and othercoagulation proteases. However, the grafting method required along reaction time and a rigorous reaction process [12–14], andits use to modify hollow fiber membranes is difficult.
Blending, in contrast, is the simplest method to modify PES bothflat sheet and hollow fiber membranes, and thus is widely used inindustry. By directly blending hydrophilic polymers, such as poly-(vinyl pyrrolidone) (PVP) [15], polyethylene glycol (PEG) [16] andpoly(acrylic acid–co-vinyl pyrrolidone) (PAA-co-PVP) [17,18], the
ll rights reserved.

F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381 3371
surface hydrophilicity of the membranes could be increased; theanti-fouling properties and blood compatibility were also im-proved [19]. PVP is a non-ionic water soluble polymer and exhibitsmany fascinating properties [20,21]. It was initially used as a bloodplasma substitute and later has been applied in a wide variety ofapplications, such as biomaterials and coatings, contact lenses, dis-infectants, intra-ocular lenses, medicine, pharmacy, cosmetics andindustrial production [22,23]. PVP has been used as a hydrophilicadditive and membrane pore-forming agent to modify PES mem-branes [24,25].
However, hydrophilic polymers such as PVP and PAA-co-PVPare water soluble and elution of the polymers from the PES mem-branes is unavoidable [26]. Thus a novel hydrophilic silica–PVPnanocomposite additive was synthesized [27], and used to improvethe surface coverage of PVP on PES membrane surfaces to enhancethe anti-fouling properties. The results indicated that the anti-fouling ability of a PES membrane with a silica–PVP nanocompos-ite additive was better than that of a PES membrane with a PVPadditive. To avoid elution acrylonitrile (AN) and methyl methacry-late (MMA) were used to synthesize co-polymers containing vinylpyrrolidone (VP) chains by free radical solution polymerization inrecent studies [28,29]; the co-polymers poly(acrylonitrile–co-acrylic acid–co-vinyl pyrrolidone) P(AN-co-AA-co-VP) [28] andpoly(methyl methacrylate–co-acrylic acid–co-vinyl pyrrolidone)P(MMA-co-AA-co-VP) [29] were blended with PES to prepare themodified membranes. The terpolymers were water insoluble dueto the AN (or MMA) chains. The acrylic acid (AA) and VP chainsare hydrophilic. The terpolymers can be directly blended withPES as macromolecular additives using N-methyl-2-pyrrolidone(NMP) as the solvent to prepare the membranes. When theterpolymers were blended in the membranes protein adsorptiondecreased while the protein anti-fouling properties increased.
The PVP-based co-polymer effectively improved the hydrophilic-ity and anti-fouling properties of the PES membranes. However, PVPand its co-polymers are synthesized by conventional free-radicalpolymerization techniques [30] and the need for further control overthe constitution and functionality of polymeric materials, accessiblethrough facile and general syntheses, remains a challenge in thepolymer sciences, to prepare a regular structure block co-polymer.In fact, VP cannot easily undergo living/controlled polymerization,since it does not form stable radicals. In the past ingenuous alterna-tive routes were explored to overcome this issue [31]. There aresome recent reports referring to living radical polymerization ofVP and the preparation of VP-based block co-polymers. Hadjichristi-dis et al. [32] reported that both nitroxide-mediated radical poly-merization (NMRP) and addition–fragmentation chain transfer(RAFT) polymerization techniques could be used for the controllableliving polymerization of VP. In order to prepare PVP block co-poly-mers they combined anionic polymerization and NMRP. The co-polymers had relatively broad molecular weight distributions [33].Recently Yamago et al. [34] reported highly controlled living radicalpolymerization of VP. Based on organostibine-mediated living radi-cal polymerization, PVP with the expected number average molecu-lar weight was synthesized. Up to now several block co-polymers,such as poly(N-isopropylacrylamide–co-hydroxylethyl methacry-late)–b-poly(vinyl pyrrolidone) (P(NIPAAm-co-HEMA)-b-PVP)[35], poly(N-isopropylacrylamide)–b-poly(vinyl pyrrolidone) (PNI-PAAm-b-PVP) [36], poly(vinyl pyrrolidone)–b-poly(methacryloyl-N0-(a-naphthyl)-thiourea) (PVP-b-PMANTU) [37], poly(vinylpyrrolidone)–b-poly(vinyl pyrrolidone–alt-maleic anhydride)–b-poly(vinyl pyrrolidone) (PVP-b-P(VP-alt-MAn)-b-PVP) [38],poly(vinyl acetate)–b-poly(vinyl pyrrolidone) (PVP-b-PVAc) [39],poly(e-caprolactone–b-vinyl pyrrolidone) (PCL-b-PVP) [40], poly-styrene–b-poly(vinyl pyrrolidone) (PS-b-PVP) [41,42], poly(vinylpyrrolidone)–b-poly(styrene–alt-maleic anhydride) (PVP-b-PSMA)[43], poly(vinyl prrolidone)–b-poly(N,N-dimethylaminoethyl
methacrylate) (PVP-b-PDMAEMA) [44] and poly(vinyl pyrroli-done)–b-poly(D,L-lactide) (PVP-b-PDLLA) [45] have been synthe-sized by anionic polymerization, atom transfer radicalpolymerization (ATRP), RAFT, and NMRP. Micelles formed fromthese co-polymers were investigated for application as drug deliverysystems. It is a great pity that none of the above research involveddetermining the biocompatibility of the products [46].
RAFT polymerization is tolerant of a wide range of functional-ities on the monomer and solvent and can effectively control therate and degree of polymerization [47–49]. In this paper the poly-merization of VP was controlled by RAFT polymerization using car-boxyl-terminated trithiocarbonate as the RAFT agent. PMMA,which is often used as a biomaterial and medical material, wasintroduced as a hydrophobic block to the macro-RAFT agent PVPto synthesize the amphiphilic triblock co-polymer poly(vinyl pyr-rolidone)–b-poly(methyl methacrylate)–b-poly(vinyl pyrrolidone)(PVP-b-PMMA-b-PVP) for the first time. The block co-polymerwas used as a macromolecular additive for direct blending withPES to prepare permanently hydrophilic PES membranes, and thecontact angles, protein adsorption, blood compatibility (plateletadhesion and clotting time) and cytocompatibility (cell culture, cellmorphology and MTT assay) of the membranes were investigated.In addition, block co-polymer modified PES hollow fiber mem-branes were prepared, and ultrafiltration experiments were carriedout to study the anti-fouling properties of the blended hollow fibermembranes.
2. Materials and methods
2.1. Materials
PES (Ultrason E6020P) was obtained from BASF, Germany. MMA(99.0% pure) was purchased from Uni-Chem, China. VP (99.0%pure) and tetrabutylammonium hydrogen sulfate were purchasedfrom Alfa Aesar, USA. VP was pretreated with activated carbon be-fore use. N,N-Dimethylacetamide (DMAC) (AR, 99.0% pure) andN,N-dimethylformamide (DMF) (99.0% pure) were purchased fromChengdu Kelong Inc. (Chengdu, China) and used as the solvents.Azo-bis-isobutryonitrile (AIBN) was purchased from Chengdu Ke-long Inc., and 4,40-azo-bis(4-cyanovaleric acid) (ACVA) was pur-chased from Alfa Aesar China (Tianjin, China), both of whichwere used as initiators. Bovine serum albumin (BSA) fraction Vand 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bro-mide (MTT) were purchased from Sigma, USA. All other chemicals(analytical grade) were obtained from Chengdu Kelong Inc., andwere used without further purification.
2.2. Synthesis and characterization of poly(vinyl pyrrolidone)–b-poly(methyl methacrylate)–b-poly (vinyl pyrrolidone)
2.2.1. RAFT agentAccording to the literature [50] carbon disulfide (27.4 g,
0.36 mol), chloroform (107.5 g, 0.9 mol), acetone (52.3 g, 0.9 mol),and tetrabutylammonium hydrogen sulfate (2.41 g, 7.1 mmol)should be mixed with 120 ml of mineral spirit in a 1 l jacketedreactor cooled with tap water under nitrogen. Sodium hydroxide(50%) (201.6 g, 2.52 mol) was added dropwise over 90 min tomaintain the temperature below 25 �C. The reaction was carriedout overnight. Then 900 ml of water was added to dissolve the so-lid, followed by 120 ml of concentrated HCl to acidify the aqueouslayer, and then the mixture was stirred for 30 min with nitrogenpurging. After filtering and thoroughly rinsing with water the solidwas dried to constant weight. 41.3 g of earth colored product wascollected.

3372 F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381
Characterization. 1H NMR (DMSO-d6, p.p.m. from TMS): 1.59 (s,12H, –CH3), 12.91 (s, 2H, –COOH). Fourier transform infrared (FTIR)spectrum (KBr, cm–1): 1710 (s, m>C@O), 1060 (s, m>C@S).
2.2.2. Macro-RAFT agent (PVP) (see step 1 in Scheme 1)Polymerization of VP was carried out in a sealed tube. The gen-
eral procedure is as follows. VP, the RAFT agent, ACVA, and H2Owere added to a tube. After bubbling with nitrogen for 30 minthe reaction mixture was allowed to warm to 80 �C under a nitro-gen atmosphere, and polymerization was carried out for 5 h. Afterprecipitation in ethyl ether the product, PVP, was dried under vac-uum at 50 �C overnight.
Characterization. 1H NMR (DMSO-d6, p.p.m. from TMS): 12.91 (s,2H), 1.59 (s, 12H) for the RAFT agent terminated segment, and 3.51(s, H, –CH–N), 3.13 (s, 2H, –CH2–N), 2.20 (s, 2H, –CH2–C@O), 2.04(s, 2H, –CH2–C–C@O), 1.86 (s, 2H, –CH2–C–N) for PVP. FTIR spectra(KBr, cm–1): 1060 (s, m>C@S) for the RAFT agent terminated segment,and 1668.2 (s, m>C@O) for PVP.
2.2.3. Amphiphilic triblock co-polymer (PVP-b-PMMA-b-PVP) (see step2 in Scheme 1)
Co-polymerization of MMA with PVP was carried out in a sealedtube. The general procedure is as follows. MMA, the macro-RAFTagent (PVP), AIBN, and DMF were added to a tube. After bubblingwith nitrogen for 30 min the reaction mixture was allowed towarm to 80 �C under a nitrogen atmosphere and polymerizationwas carried out for 5 h. After precipitation in ethyl ether the prod-uct was dried under vacuum at 50 �C overnight. The product ob-tained was ground to a fine powder and immersed in water andTHF for 1 week each. This was repeated three times, and then theproduct was dried under vacuum at room temperature for 24 h.
Scheme 1. Synthesis of the PVP-b-PMMA-b-PVP block co-polymer.
Characterization. 1H NMR (DMSO-d6, p.p.m. from TMS): 12.91 (s,2H), 1.59 (s, 12H) for the RAFT agent terminated segment, 3.51 (s,H, –CH–N), 3.13 (s, 2H, –CH2–N), 2.20 (s, 2H, –CH2–C@O), 2.04 (s,2H, –CH2–C–C@O), 1.86 (s, 2H, –CH2–C–N) for the PVP block, and3.32–3.56 (s, 3H, CH3–O–), 1.76 (s, 2H, –CH2–C–CH3), 0.73–0.93 (s,2H, CH3–C–C@O) for the PMMA block. FTIR spectra (KBr, cm–1):1060 (s, m>C@S) for the RAFT agent terminated segment, 1668.2 (s,m>C@O) for the PVP block, and 1734.9 (s, m>C@O) for the PMMA block.
2.2.4. CharacterizationThe FTIR spectrum of the co-polymer was measured using a FT-
IR Nicolet560 (Nicol America) instrument. To prepare FTIR samplesthe polymer was dissolved in NMP and cast on a potassium bro-mide (KBr) disk with the thickness of about 0.8 mm. The 1H NMRspectra were recorded on a Varian Unity Plus 300/54 NMR spec-trometer using DMSO-d6 as the solvent at room temperature. Theweight averaged molecular weight of PVP was measured with amulti-angle light scattering detector (BI-200SM, BrookhavenInstruments Co., Holtsville, NY) using H2O as the solvent at 25 �C.
2.3. Preparation and characterization of co-polymer blendedpolyethersulfone membranes
The PES/co-polymer membranes were prepared by a phaseinversion technique. PES and the synthesized co-polymer were dis-solved in the solvent DMAC by vigorous stirring until a clear homo-geneous solution was obtained. The concentration of PES was16 wt.%). In the experiments different membranes were preparedby changing the weight percentage of the co-polymer in the cast-ing solutions. The content of co-polymer in the casting solutionswas 0, 1, 3, and 5 wt.% (the maximum amount). After vacuumdegassing the casting solutions were prepared as membranes byspin coating coupled with a liquid–liquid phase separation tech-nique at room temperature. The membranes were thoroughlyrinsed with distilled water to remove the residual solvent. All theprepared membranes with PES/co-polymer ratios of 16/0, 16/1,16/3, and 16/5 (wt.%) were of uniform thickness of about 60–70 lm, and were termed FSM-0, FSM-1, FSM-3 and FSM-5,respectively.
Thermogravimetric analysis (TGA) (TA-500) was used to inves-tigate the changes in thermal stability of the co-polymer and mod-ified membranes at a heating rate of 10 �C min–1 under a dry N2
atmosphere. The morphologies of the membranes were observedby scanning electron microscopy (SEM) using an XL 30ESME scan-ning microscope. The membranes, frozen in liquid nitrogen, werebroken and sputtered with a gold layer before SEM analysis. Thestructures and elements of the membrane surfaces were investi-gated by reflected FTIR and X-ray photoelectron spectroscopy(XPS).
The hydrophilicity of the membrane surface was characterizedon the basis of contact angle measurements using a contact anglegoniometer (OCA20, Dataphysics, Germany) equipped with videocapture. A piece of membrane 2 � 2 cm was attached to a glassslide and mounted in the goniometer. For the static contact anglemeasurements 3 ll double distilled water was dropped onto thesurface of the membrane at room temperature and the contact an-gle was measured after 10 s. At least eight measurements wereaveraged to obtain a reliable value. The measurement error was±3�.
2.4. Ultrafiltration and anti-fouling properties of the modifiedpolyethersulfone hollow fibers
2.4.1. Preparation of the hollow fiber membrane and filterPES/co-polymer hollow fiber membranes were prepared using
DMAC as the solvent. The PES/co-polymer solution (the contents

F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381 3373
of PES and co-polymer were 16 and 5 wt.%, respectively) was de-gassed to remove air bubbles and then a dry–wet spinning tech-nique was used to fabricate the PES hollow fiber membranes[29,51]. The membranes were immersed in water for >24 h to elutethe residual DMAC, with the extraction water being changed every3 h. Then the membranes were placed in 50 wt.% aqueous glycerolsolution for 24 h to prevent collapse of the porous structures whenthey were dried. The resultant fibers were then dried in air at roomtemperature. The hollow fiber membrane filters were prepared byemploying epoxy resin as the potting material, with an effectivearea of about 120 cm2. The prepared membranes with PES/co-polymer ratios of 16/0 and 16/5 are termed HFM-0 and HFM-5.
2.4.2. Ultrafiltration of the PEG solutionThe permeability of the hollow fiber membranes was investi-
gated using polyethylene glycol (PEG-10000). The feed solutionswere prepared by dissolving PEG-10000 in double distilled waterat a concentration of 0.1 g l–1. The PEG-10000 solution was appliedto the membrane using the apparatus described in our earlierstudy [52]. The permeant solution was collected and the flux calcu-lated using Eq. (1).
Fluxðml=m2 �mmHg � hÞ�1 ¼ V=StP ð1Þ
where V is the volume of the permeant solution, S is the effectivemembrane area, t is the time of solution collection, and P is thepressure applied to the hollow fiber membrane. The concentrationof PEG was determined using a UV–VIS spectrophotometer (model756, Shanghai Spectrophotometer Instrument Co., Shanghai, China)at a wavelength of 510 nm after reaction with Ninhydin. The ob-served sieving coefficient (S0) of PEG was defined as follows:
S0 ¼ Cp=Cb ð2Þ
where Cp is the permeant concentration of PEG and Cb is the bulkconcentration.
2.4.3. Ultrafiltration of the protein solutionUltrafiltration of bovine serum albumin (BSA) solution was car-
ried out to study the anti-fouling properties of the modified PEShollow fiber membranes. BSA solution was prepared by dissolvingBSA in phosphate-buffered saline (PBS), adjusted to pH 7.4 withhydrochloric acid, at a concentration of 1.0 mg ml–1. The hollow fi-bers under test were pre-compacted at an internal pressure of135 mm Hg and external pressure of 100 mm Hg for 30 min underPBS flow to obtain a steady state, then the internal pressure was re-duced to 70 mm Hg and the external pressure was reduced to40 mm Hg, and the PBS flux was measured. After 30 min filtrationthe feed solution was switched to 1.0 mg ml–1 BSA solution (pH7.4) and the flux again measured. Finally, the filter and the solutionreservoir were fully emptied and refilled with deionized water. Thehollow fiber membranes were washed with deionized water for30 min. Then the PBS solution flux was again measured. The fluxesof PBS and BSA solution through the hollow fibers were calculatedusing Eq. (1).
In order to evaluate the fouling-resistance ability of the mem-branes the flux recovery ratio (FRR) was calculated using theexpression:
FRR ð%Þ ¼ ðF2=F1Þ � 100 ð3Þ
where F1 and F2 (ml/(m2 mmHg h)–1) are the PBS solution flux be-fore and after the protein ultrafiltration experiment, respectively.
2.5. Blood compatibility
2.5.1. Protein adsorptionThe protein adsorption experiments were carried out with BSA
solution (1 mg ml–1 in PBS, pH 7.4). The membrane, with an area of1 � 1 cm, was incubated in PBS for 24 h and then immersed in theprotein solution for 2 h at 37 �C. After protein adsorption the mem-branes were gently rinsed with PBS and then immersed in glasstubes containing 2 wt.% aqueous sodium dodecyl sulfate (SDS)solution for 1 h at 37 �C to remove the proteins adsorbed ontothe membranes. The amount of protein eluted into the SDS solu-tion was quantified with a Micro BCA™ protein assay reagent kit.More than 95% of the adsorbed protein could be eluted into theSDS solution. Then the amount of adsorbed BSA was calculated.
2.5.2. Platelet adhesionHealthy fresh human blood (male, 25 years old) was collected
using vacuum tubes containing sodium citrate as an anti-coagu-lant; the concentration of sodium citrate was 3.8 wt.%, the ratioof anticoagulant to blood was 10:90 vol.%. The blood was centri-fuged at 1500 r.p.m. for 15 min to obtain platelet-rich plasma(PRP) or at 4000 r.p.m. for 15 min to obtain platelet-poor plasma(PPP).
The PES and modified PES membranes were immersed in PBSand equilibrated at 37 �C for 1 h. The PBS was removed and then1 ml of fresh PRP was introduced. The membranes were incubatedwith PRP at 37 �C for 2 h. Then the PRP was decanted off and themembranes were rinsed three times with PBS. Finally, the mem-branes were treated with 2.5 wt.% glutaraldehyde in PBS at 4 �Cfor 1 day. The samples were washed with PBS, subjected to adrying process by passing them through a series of graded alco-hol–PBS solutions (25%, 50%, 75%, and 100%) and isoamyl ace-tate–alcohol solutions (25%, 50%, 75%, and 100%). Plateletadhesion was observed using an S-2500C microscope (Hitachi,Japan). The number of adherent platelets on the membranes wascalculated from five SEM pictures at 500 �magnification from dif-ferent places on the same membrane.
2.5.3. Clotting timeTo evaluate the anti-thrombogenicity of the membranes the
activated partial thromboplastin time (APTT) was measured withan automated blood coagulation analyzer CA-50 (Sysmex Corp.,Kobe, Japan). To measure APTT samples (1 � 1 cm, one piece) wereincubated with 0.15 ml of healthy human blood plasma at 37 �C for30 min and then the APTT was measured.
2.6. Cytocompatibility
2.6.1. Cell cultureHuman embryonic hepatocytes (LO-2) were grown in R1640
medium supplemented with 10% fetal bovine serum (FBS)(Hyclone, USA), 2 mM L-glutamine and 1 vol.% antibiotics mixture(10,000 U penicillin and 10 mg streptomycin). Cultures were main-tained in a humidified atmosphere of 5% CO2 at 37 �C (Queue Incu-bator, Paris, France). Confluent cells were detached from theculture flask with sterilized PBS and 0.05% trypsin/EDTA solution.The culture medium was changed every day.
The PES and modified PES membranes were cut into 1 � 1 cm tofit 24-well cell culture polystyrene plates and pre-wetted byimmersion in culture medium for 3 h in a 37 �C incubator. Thenthe membranes were placed in the cell culture plates, rinsed withPBS and sterilized by c-irradiation.
2.6.2. Cell morphology on the membranesFor SEM observation hepatocytes were seeded onto the mem-
branes at a density of approximately 2.5 � 104 cells cm–2. After

3374 F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381
4 days the seeded membranes were rinsed with PBS and fixed with2.5 wt.% glutaraldehyde in PBS at 4 �C for 12 h. For morphologyobservation the fixed samples were subjected to a drying processby passing them through a series of graded alcohol–PBS solutions(30%, 50%, 70%, 80%, 90%, 95%, and 100%, 15 min each) and thendehydrated through isoamyl acetate. Critical point drying of thespecimens was carried out with liquid CO2. The specimens weresputter coated with a gold layer and examined in an S-2500Cmicroscope (Hitachi, Japan).
2.6.3. MTT assayAfter cell culture for 2, 4, and 7 days viability of the hepatocytes
was determined by MTT assay. The hepatocytes were seeded ontothe membranes at a density of approximately 2.5 � 104 cells cm–2.Cells cultured in wells without membranes served as controls inthis study. After predetermined time intervals 45 ll of MTT solu-tion (1 mg ml–1 in the test medium) was added to each well andincubated for 4 h at 37 �C. Mitochodrial dehydrogenases of viablecells selectively cleave the tetrazolium ring, yielding blue/purpleformazan crystals. Then 400 ll of ethanol was added to dissolvethe formazan crystals. Thus the quantity of formazan dissolved inthe ethanol reflects the level of cell metabolism. The solutionwas shaken homogeneously for about 15 min. The sample solu-tions were aspirated into microtiter plates and the optical densityread in a Microplate reader (model 550, Bio-Rad) at 492 nm. Allexperiments were repeated three times, and the results are ex-pressed as means ±SD. The statistical significance was assessedby Student’s t-test, with the level of significance set at P < 0.05.
3. Results and discussion
3.1. Synthesis and characterization of poly(vinyl pyrrolidone)–b-poly(methyl methacrylate)–b-poly(vinyl pyrrolidone) amphiphilictriblock co-polymer
The schematic procedure for the synthesis of PVP-b-PMMA-b-PVP is shown in Scheme 1. As shown in the scheme, the co-polymer retained the carboxyl-terminated structure of the RAFTagent, which helps to improve, or at least not decrease, the amph-iphilicity of the co-polymer compared with other RAFT agents [53].For the facile polymerization of VP trithiocarbonate was selected asthe RAFT agent, and the PVP synthesized was further used as amacro-RAFT agent for the co-polymerization of MMA with PVP.The chain transfer reaction between the propagation radical andthe macro-RAFT agent happens randomly and identically. That isto say the MMA or PMMA propagation chains insert not only inthe left side between the PVP (M1) and the S atom but also inthe right side between the PVP (M1) and the S atom, resulting inABBA or ABA block co-polymers rarely containing fractions of di-block species. Meanwhile, it is expected that more PMMA homo-polymer will be produced than PVP homopolymer. To extract thehomopolymers of PVP and PMMA which are probably producedby bi-radical termination the prepared product was ground to afine powder and immersed in water and THF for 1 week each,
Table 1Composition and molecular weight data of the copolymer.a
Feed ratio (PVP wt.%) Calculated (NMR) (PVP wt.%)b
25 19.5
a The polymerization was conducted at 80 �C for 12 h using DMF as the solvent.b Estimated from the 1H-NMR Spectrum of the copolymer.c Weight-average molecular weight measured by multi angle light scattering detectod Molecular weight calculated by comparing the features peaks of PMMA methyl (1.0
which was repeated three times alternately. The remaining whitepowder was the triblock co-polymer of PVP-b-PMMA-b-PVP.
The structure of the purified PVP-b-PMMA-b-PVP triblock co-polymer was demonstrated by FTIR and 1H NMR, and is specifiedin Section 2.2.3. The co-polymer compositions and molecularweights, determined from the 1H NMR spectra and multi-anglelight scattering, are summarized in Table 1. The PVP content(wt.%) estimated from 1H NMR spectra and the amount added be-fore polymerization were almost identical. On the basis of the ratioof the signal intensities of the peaks at d 1.0–0.51 p.p.m. to those atd 3.25–2.91 p.p.m. the degrees of polymerization of the PVP andPMMA blocks were about 62 and 285. In addition, according to aprevious study, it is easy to control the molecular weights andcompositions of the co-polymers by changing the molecularweight of PVP or the ratios of MMA to PVP, and thus to design dif-ferent well-defined triblock co-polymers for biomedical applica-tions [54].
3.2. Membrane preparation and characterization
PVP-b-PMMA-b-PVP modified PES membranes were preparedby a liquid–liquid phase separation technique at room temperaturewith 16% PES and 0–5% block co-polymer. In order to evaluate thecomposition of the modified membrane the thermal degradation ofthe co-polymer PVP-b-PMMA-b-PVP and PES/PVP-b-PMMA-b-PVP(16/5) membranes was determined by TGA. As shown in Fig. 1A,the modified PVP-b-PMMA-b-PVP/PES membrane had two obviousweight loss peaks, below about 450.3 �C, where the co-polymer de-graded, and above about 450.3 �C, where the PES matrix degraded.From the relative weight losses it could be calculated that the ratioof the co-polymer to PES was about 1:3.8, which was close to thefeed ratio of 5:16. These studies provide evidence that the modify-ing additive PVP-b-PMMA-b-PVP was incorporated into the PESmembrane, with almost no elution during the membrane prepara-tion and processing procedures.
Fig. 1B shows cross-sectional SEM micrographs of the PES andPES/PVP-b-PMMA-b-PVP membranes. The characteristic morphol-ogy of asymmetric membranes, consisting of a dense top layerand porous bottom layer with a finger-like structure, was observedfor all membranes [55,56]. However, the porous bottom layer forthe PES/PVP-b-PMMA-b-PVP membrane was more establishedand obvious than that of the PES membrane. For the modifiedmembrane there were large numbers of micropores on the surfaceof the macrovoids, and a few self-assembled microspheres wereobserved, embedded in the pores. The SEM images suggest thatthe structure of the modified membrane had been altered by theblock co-polymer additive.
To understand how the block co-polymer is distributed insidethe PES membrane and why the structure is formed during thephase separation process the mechanism of formation can be sum-marized as follows. The initial solution, including PES, the co-polymer, and DMAC, formed a homogeneous phase, with theco-polymer macromolecules homogeneously dissolved in the solu-tion. Because of the amphiphilic nature of the block co-polymer[57,58], when phase separation began PVP-b-PMMA-b-PVP rose
Mw PVPc (�10�3) Mn copolymer
d (�10�3)
6.9 35.4
r using H2O as solvent at 25 �C.0–0.51 ppm) and PVP methylene (3.25–2.91 ppm) from 1H-NMR spectrum.
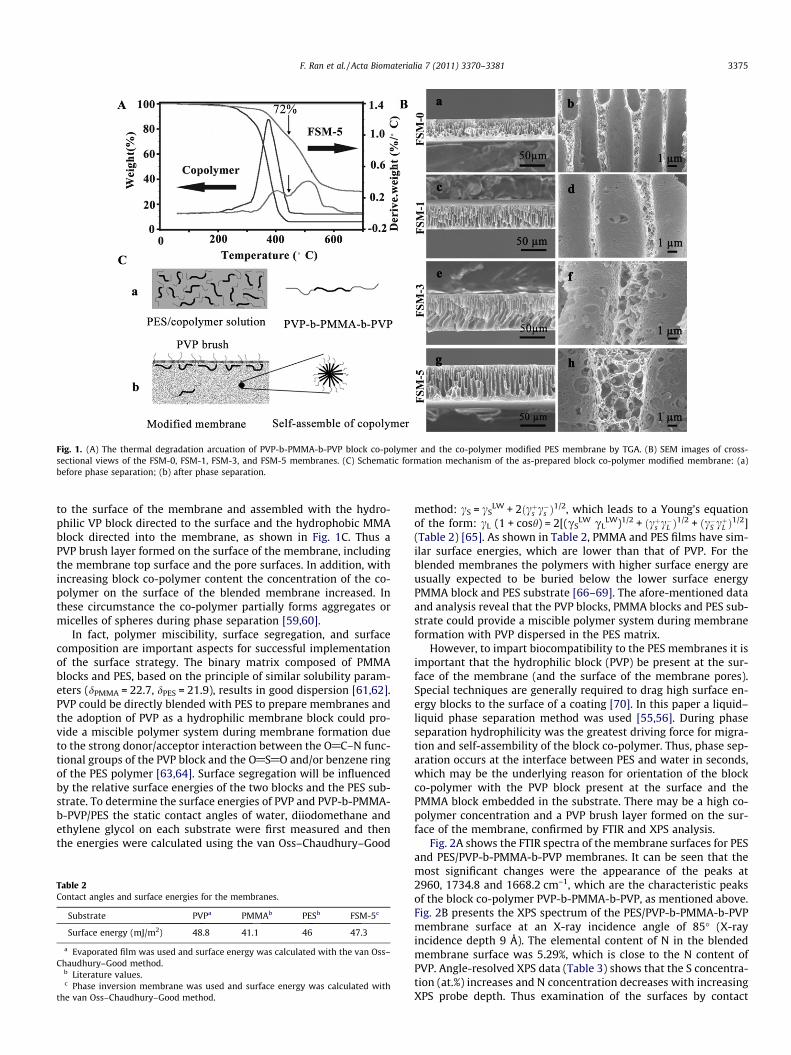
Fig. 1. (A) The thermal degradation arcuation of PVP-b-PMMA-b-PVP block co-polymer and the co-polymer modified PES membrane by TGA. (B) SEM images of cross-sectional views of the FSM-0, FSM-1, FSM-3, and FSM-5 membranes. (C) Schematic formation mechanism of the as-prepared block co-polymer modified membrane: (a)before phase separation; (b) after phase separation.
F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381 3375
to the surface of the membrane and assembled with the hydro-philic VP block directed to the surface and the hydrophobic MMAblock directed into the membrane, as shown in Fig. 1C. Thus aPVP brush layer formed on the surface of the membrane, includingthe membrane top surface and the pore surfaces. In addition, withincreasing block co-polymer content the concentration of the co-polymer on the surface of the blended membrane increased. Inthese circumstance the co-polymer partially forms aggregates ormicelles of spheres during phase separation [59,60].
In fact, polymer miscibility, surface segregation, and surfacecomposition are important aspects for successful implementationof the surface strategy. The binary matrix composed of PMMAblocks and PES, based on the principle of similar solubility param-eters (dPMMA = 22.7, dPES = 21.9), results in good dispersion [61,62].PVP could be directly blended with PES to prepare membranes andthe adoption of PVP as a hydrophilic membrane block could pro-vide a miscible polymer system during membrane formation dueto the strong donor/acceptor interaction between the O@C–N func-tional groups of the PVP block and the O@S@O and/or benzene ringof the PES polymer [63,64]. Surface segregation will be influencedby the relative surface energies of the two blocks and the PES sub-strate. To determine the surface energies of PVP and PVP-b-PMMA-b-PVP/PES the static contact angles of water, diiodomethane andethylene glycol on each substrate were first measured and thenthe energies were calculated using the van Oss–Chaudhury–Good
Table 2Contact angles and surface energies for the membranes.
Substrate PVPa PMMAb PESb FSM-5c
Surface energy (mJ/m2) 48.8 41.1 46 47.3
a Evaporated film was used and surface energy was calculated with the van Oss–Chaudhury–Good method.
b Literature values.c Phase inversion membrane was used and surface energy was calculated with
the van Oss–Chaudhury–Good method.
method: cS = cSLW + 2ðcþs c�s Þ
1/2, which leads to a Young’s equationof the form: cL (1 + cosh) = 2[(cS
LW cLLW)1/2 + ðcþs c�L Þ
1/2 + ðc�S cþL Þ1/2]
(Table 2) [65]. As shown in Table 2, PMMA and PES films have sim-ilar surface energies, which are lower than that of PVP. For theblended membranes the polymers with higher surface energy areusually expected to be buried below the lower surface energyPMMA block and PES substrate [66–69]. The afore-mentioned dataand analysis reveal that the PVP blocks, PMMA blocks and PES sub-strate could provide a miscible polymer system during membraneformation with PVP dispersed in the PES matrix.
However, to impart biocompatibility to the PES membranes it isimportant that the hydrophilic block (PVP) be present at the sur-face of the membrane (and the surface of the membrane pores).Special techniques are generally required to drag high surface en-ergy blocks to the surface of a coating [70]. In this paper a liquid–liquid phase separation method was used [55,56]. During phaseseparation hydrophilicity was the greatest driving force for migra-tion and self-assembility of the block co-polymer. Thus, phase sep-aration occurs at the interface between PES and water in seconds,which may be the underlying reason for orientation of the blockco-polymer with the PVP block present at the surface and thePMMA block embedded in the substrate. There may be a high co-polymer concentration and a PVP brush layer formed on the sur-face of the membrane, confirmed by FTIR and XPS analysis.
Fig. 2A shows the FTIR spectra of the membrane surfaces for PESand PES/PVP-b-PMMA-b-PVP membranes. It can be seen that themost significant changes were the appearance of the peaks at2960, 1734.8 and 1668.2 cm–1, which are the characteristic peaksof the block co-polymer PVP-b-PMMA-b-PVP, as mentioned above.Fig. 2B presents the XPS spectrum of the PES/PVP-b-PMMA-b-PVPmembrane surface at an X-ray incidence angle of 85� (X-rayincidence depth 9 Å). The elemental content of N in the blendedmembrane surface was 5.29%, which is close to the N content ofPVP. Angle-resolved XPS data (Table 3) shows that the S concentra-tion (at.%) increases and N concentration decreases with increasingXPS probe depth. Thus examination of the surfaces by contact
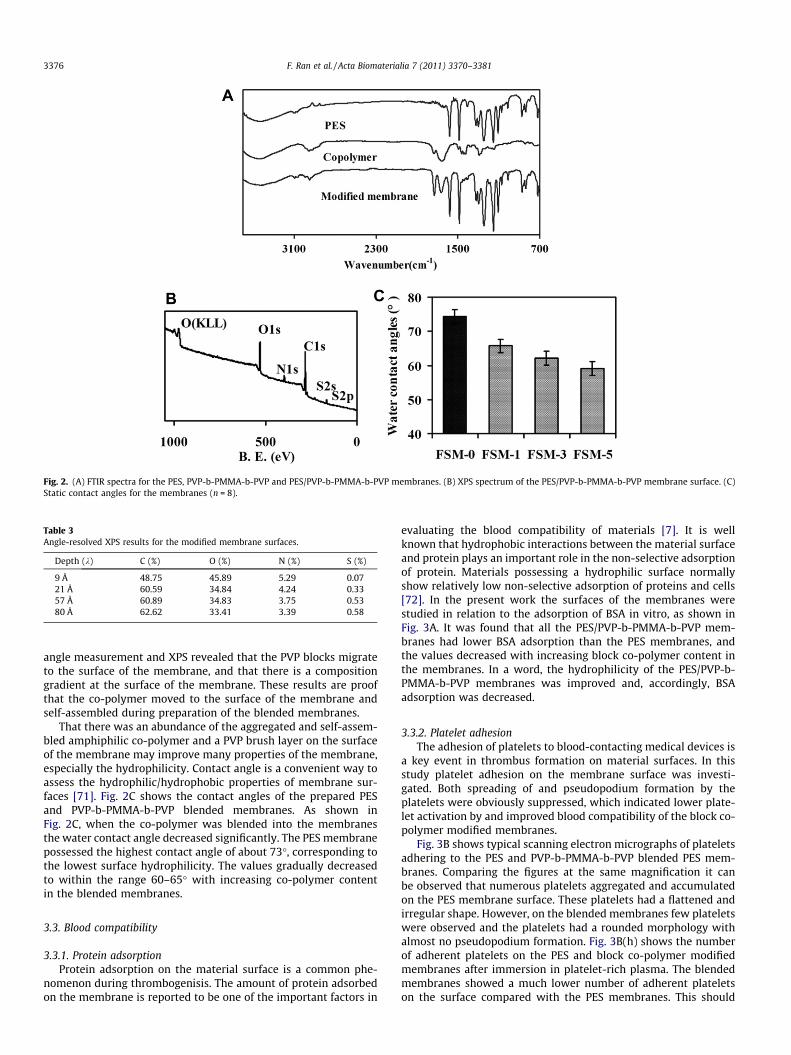
Fig. 2. (A) FTIR spectra for the PES, PVP-b-PMMA-b-PVP and PES/PVP-b-PMMA-b-PVP membranes. (B) XPS spectrum of the PES/PVP-b-PMMA-b-PVP membrane surface. (C)Static contact angles for the membranes (n = 8).
Table 3Angle-resolved XPS results for the modified membrane surfaces.
Depth (k) C (%) O (%) N (%) S (%)
9 Å 48.75 45.89 5.29 0.0721 Å 60.59 34.84 4.24 0.3357 Å 60.89 34.83 3.75 0.5380 Å 62.62 33.41 3.39 0.58
3376 F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381
angle measurement and XPS revealed that the PVP blocks migrateto the surface of the membrane, and that there is a compositiongradient at the surface of the membrane. These results are proofthat the co-polymer moved to the surface of the membrane andself-assembled during preparation of the blended membranes.
That there was an abundance of the aggregated and self-assem-bled amphiphilic co-polymer and a PVP brush layer on the surfaceof the membrane may improve many properties of the membrane,especially the hydrophilicity. Contact angle is a convenient way toassess the hydrophilic/hydrophobic properties of membrane sur-faces [71]. Fig. 2C shows the contact angles of the prepared PESand PVP-b-PMMA-b-PVP blended membranes. As shown inFig. 2C, when the co-polymer was blended into the membranesthe water contact angle decreased significantly. The PES membranepossessed the highest contact angle of about 73�, corresponding tothe lowest surface hydrophilicity. The values gradually decreasedto within the range 60–65� with increasing co-polymer contentin the blended membranes.
3.3. Blood compatibility
3.3.1. Protein adsorptionProtein adsorption on the material surface is a common phe-
nomenon during thrombogenisis. The amount of protein adsorbedon the membrane is reported to be one of the important factors in
evaluating the blood compatibility of materials [7]. It is wellknown that hydrophobic interactions between the material surfaceand protein plays an important role in the non-selective adsorptionof protein. Materials possessing a hydrophilic surface normallyshow relatively low non-selective adsorption of proteins and cells[72]. In the present work the surfaces of the membranes werestudied in relation to the adsorption of BSA in vitro, as shown inFig. 3A. It was found that all the PES/PVP-b-PMMA-b-PVP mem-branes had lower BSA adsorption than the PES membranes, andthe values decreased with increasing block co-polymer content inthe membranes. In a word, the hydrophilicity of the PES/PVP-b-PMMA-b-PVP membranes was improved and, accordingly, BSAadsorption was decreased.
3.3.2. Platelet adhesionThe adhesion of platelets to blood-contacting medical devices is
a key event in thrombus formation on material surfaces. In thisstudy platelet adhesion on the membrane surface was investi-gated. Both spreading of and pseudopodium formation by theplatelets were obviously suppressed, which indicated lower plate-let activation by and improved blood compatibility of the block co-polymer modified membranes.
Fig. 3B shows typical scanning electron micrographs of plateletsadhering to the PES and PVP-b-PMMA-b-PVP blended PES mem-branes. Comparing the figures at the same magnification it canbe observed that numerous platelets aggregated and accumulatedon the PES membrane surface. These platelets had a flattened andirregular shape. However, on the blended membranes few plateletswere observed and the platelets had a rounded morphology withalmost no pseudopodium formation. Fig. 3B(h) shows the numberof adherent platelets on the PES and block co-polymer modifiedmembranes after immersion in platelet-rich plasma. The blendedmembranes showed a much lower number of adherent plateletson the surface compared with the PES membranes. This should

Fig. 3. (A) Protein adsorption by the modified membranes. (B) SEM images of platelets adhering to the membranes (h, number of the adherent platelets on the membranesadsorbed from platelet-rich plasma estimated from the SEM pictures). (C) Activated partial thromboplastin time (APTT) for the modified PES membranes.
F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381 3377
be attributed to the difference in the PVP brush layer on the surfaceof the modified membranes.
3.3.3. Clotting timeTo further study the blood compatibility of the modified mem-
branes the APTT was measured, as shown in Fig. 3C. The APTT testdetermines the bioactivity of intrinsic blood coagulation factors. Itwas found that the APTT of the block co-polymer blended mem-branes increased compared with the PES membrane, and withincreasing block co-polymer content the APTT of the modifiedmembranes increased, being nearly twice that of the PES mem-brane at 5% co-polymer. The enhancement in anticoagulant activ-ity might have resulted from partial contributions of surfacehydrophilicity, lower protein adsorption, and depressed plateletadhesion and activation.
Compared with others methods to improve the blood compati-bility of bulk materials, such as grafting heparin and insulin ontopoly(ethylene terephthalate) films [73] or grafting poly(ethyleneglycol) onto silicon and glass surfaces [74], the enhancement ofAPTT after grafting was lower than that of the block co-polymermodified membranes produced in this study. However, Lin et al.[75] reported that the enhancement of APTT increased sharplywith increasing graft density of heparin on a polyacrylonitrile sur-face. Nevertheless, grafting, in contrast to blending, is a complexmethod to modify bulk materials, especially hollow fibers, and ishard to use in an industrial setting. Recently Wang et al. [19] re-ported that blending PVP as an additive in a PES membrane im-proved the blood compatibility, and the APTT increasedsignificantly. However, the elution of PVP was unavoidable. Onthe other hand, compared with others studies [75], almost noadherent platelets were observed when 5% co-polymer wasblended in, as shown in Fig. 3B(h). Thus, this is a simple and effec-tive way to improve the blood compatibility of the matrix.
As discussed above, the modified PES membrane showed goodblood compatibility. As we know, most of the membranes used
in blood purification are hollow fiber type membranes. To provideinformation on real application of the modified PES membranes inblood purification co-polymer modified hollow fiber PES mem-branes were prepared, the permeability and protein anti-foulingproperties of which are discussed below.
3.4. Ultrafiltration and antifouling properties of modified PES hollowfibers
3.4.1. Preparation and characterization of the hollow fiber membraneThe block co-polymer modified PES hollow fiber membrane was
spun by a dry–wet spinning method. SEM images of cross-sectionalviews of the hollow fiber membranes are shown in Fig. 4A. The pre-pared blended membranes (FSM-5) were opaque in appearanceand satisfactorily strong for high pressure applications. The wallthickness of the hollow fiber was about 110 lm, and the innerdiameter was about 400 lm. A skin layer was found on both sidesof the membrane wall, under which there was a finger-like struc-ture layer, and then a porous layer. Furthermore, it was clearly ob-served that the finger-like structure was in the middle of themembrane. This was caused by the exchange between NMP andwater during membrane formation. There were no obvious differ-ences between HFM-0 and HFM-5 except for the greater porosityof the modified membrane. This indicated that small amounts ofthe co-polymer did not influence the main structure of the PESmembrane, and the co-polymer was well incorporated into thehollow fiber membrane. A schematic of the mechanism of forma-tion of the modified membrane (Fig. 1C) also indicated that PVPbrushes could be formed on the surface of the modified hollow fi-ber membranes.
3.4.2. Permeability to PEG solutionFor end-stage renal disease patients hemodialysis is widely
used as a life sustaining treatment. However, it is difficult to re-move middle molecular weight toxins (such as b2-microglobulin

Fig. 4. (A) SEM images of cross-sectional views of the PES hollow fiber membranes.(B) Time-dependent fluxes of the modified membranes during the ultrafiltrationprocess for the PES and modified PES membranes: (a) HFM-5; (b) HFM-0. Distilledwater and protein solution were used alternately in ultrafiltration (n = 3).
3378 F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381
(b2-M)) during conventional hemodialysis. b2-M, which is consid-ered one of the middle molecular weight toxins for end-stage renaldisease patients [76], is a non-glycosylated protein with a molecu-lar weight of 11,800 Da. Free b2-M is found in the body fluids as aresult of shedding from cell surfaces or intracellular release[77,78]. Generally the content of b2-M in the body of end-stage re-nal disease patients is higher than that in a healthy one. Thus it isnecessary to remove b2-M during hemodialysis. Now, most highflux hemodialysis membranes have been designed to removeb2-M during hemodialysis.
To investigate whether the modified hollow fiber membranecould effectively remove b2-M the observed sieving coefficient(So) for polyethylene glycol (PEG) was evaluated. PEG-10000 wasselected in order to simulate the molecule weight of b2-M. PEGhas been widely used as a standard macromolecule in ultrafiltra-tion experiments to determine the sieving coefficient. The ob-served sieving coefficients for PEG-10000 of the PES andmodified hollow fiber membranes were 28.64% and 38.38%,respectively. So for the modified PES hollow fiber membrane in-creased after blending with the co-polymer. The results also sug-gest that the modified hollow fiber membranes could effectivelyremove b2-M, and can be used as high flux hemodialysismembranes.
3.4.3. Membrane anti-fouling propertiesUltrafiltration experiments were also carried out to study the
protein anti-fouling properties of the blended hollow fiber mem-branes to provide further information as to practical applications.Fig. 4B shows the time-dependent flux during ultrafiltration. As
shown in Fig. 4B, the flux recovery ratio of the modified hollow fi-bers increased from 50.6% (PES hollow fibers) to 96.6% due toblending with the amphiphilic block co-polymer.
The flux decreased, but not dramatically, in the initial stage ofBSA solution ultrafiltration due to membrane fouling caused bythe deposition and adsorption of protein molecules on the mem-brane surface and in the membrane pores. When the adsorptionof protein molecules became saturated a relatively steady fluxwas reached during the final stage of BSA solution ultrafiltration.After 30 min protein ultrafiltration the membranes were cleanedwith flowing double distilled water; PBS solution flux of the mem-branes recovered to some extent. The modified PES membranesshowed a large flux recovery ratio of nearly 100%, which couldbe explained by the fact that VP chains increase the hydrophilicityof hollow fiber membranes, passivity the membrane surface, andlower protein adsorption [79,80]. Recently Zhao et al. [81] investi-gated the anti-fouling properties of a poly(ethylene glycol)-baseddiblock co-polymer modified PES hollow fiber; the flux recoveryratio increased from 56.3% to 86.5% after blending in the diblockco-polymer. However, it was lower compared with the flux recov-ery ratio of the PVP-based triblock co-polymer modified PES hol-low fiber (96.6%) prepared in this study, which might beattributed to the PVP brush layer that formed on the surface ofthe membrane, as discussed above.
3.5. Cytocompatibility
It is well known that hollow fiber membranes have been widelyused in blood purification therapies, including plasmapheresis,hemodialysis, hemodiafiltration, and bioartificial liver support[82]. The efficiency of hollow fiber-based bioartificial livers de-pends greatly on the choice of membrane in terms of permeability,cut-off and biocompatibility [83]. Thus the cytocompatibility of themodified PES membranes was investigated. In this study LO2 hu-man hepatocytes were selected to evaluate the cytocompatibilityof the modified membranes, which could possibly be used for bio-artificial liver support.
3.5.1. Cell morphologyGenerally speaking cells will undergo morphological changes to
stabilize the cell–material interface after contacting a biomaterial.The whole process of adhesion and spreading consists of cellattachment, filopodial growth, cytoplasmic webbing, flattening ofthe cell mass and ruffling of the peripheral cytoplasm progressingin a sequential fashion [84].
Fig. 5A shows the morphology of hepatocytes cultured for4 days on the PES and modified PES membranes. It can be seen thatthe hepatocytes extended pseudopodia to adhere onto the materi-als. The number of hepatocytes on the PES membrane was least.With increasing co-polymer content the number of adherent hepa-tocytes increased. On the surfaces of the modified membranes thecells spread, with ruffling of the peripheral cytoplasm, and almostcovered the whole surface, and had been become flattened with alarger area of attachment compared with cells cultured on the PESmembrane, especially on FSM-5, which indicated that addition ofthe co-polymer could promote better cell attachment and growth.
Furthermore, spheroids of hepatocytes, formed by the rear-rangement and compaction of cell aggregates, have been advo-cated as a highly useful culture mode for hepatocytes instead ofthe traditional monolayer culture, since their tissue-like structurecould promote cell proliferation and differentiation and maintainhigher level liver-specific functions over a long period [85,86].Thus spheroids of hepatocytes are ideal for bioartificial liver sup-port. From Fig. 5A it can be observed that there were some spheresor balling, especially on FSM-3 and FSM-5, in some areas. This indi-cates that the block co-polymers could induce hepatocytes to
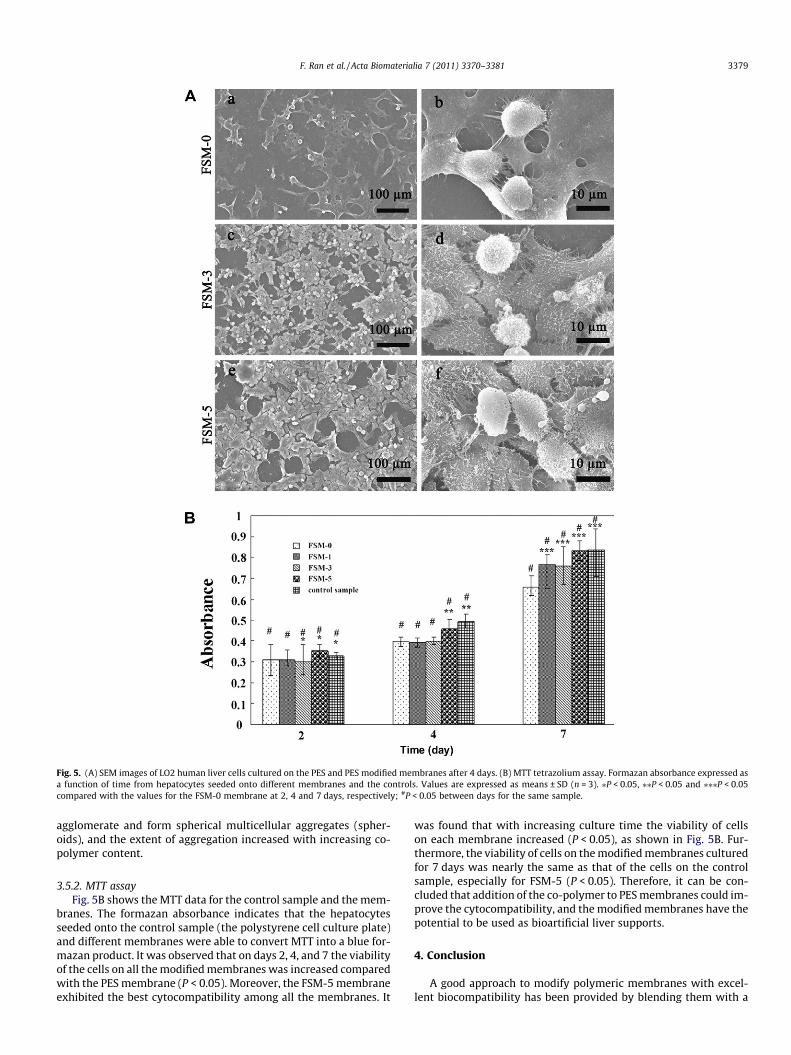
Fig. 5. (A) SEM images of LO2 human liver cells cultured on the PES and PES modified membranes after 4 days. (B) MTT tetrazolium assay. Formazan absorbance expressed asa function of time from hepatocytes seeded onto different membranes and the controls. Values are expressed as means ± SD (n = 3). ⁄P < 0.05, ⁄⁄P < 0.05 and ⁄⁄⁄P < 0.05compared with the values for the FSM-0 membrane at 2, 4 and 7 days, respectively; #P < 0.05 between days for the same sample.
F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381 3379
agglomerate and form spherical multicellular aggregates (spher-oids), and the extent of aggregation increased with increasing co-polymer content.
3.5.2. MTT assayFig. 5B shows the MTT data for the control sample and the mem-
branes. The formazan absorbance indicates that the hepatocytesseeded onto the control sample (the polystyrene cell culture plate)and different membranes were able to convert MTT into a blue for-mazan product. It was observed that on days 2, 4, and 7 the viabilityof the cells on all the modified membranes was increased comparedwith the PES membrane (P < 0.05). Moreover, the FSM-5 membraneexhibited the best cytocompatibility among all the membranes. It
was found that with increasing culture time the viability of cellson each membrane increased (P < 0.05), as shown in Fig. 5B. Fur-thermore, the viability of cells on the modified membranes culturedfor 7 days was nearly the same as that of the cells on the controlsample, especially for FSM-5 (P < 0.05). Therefore, it can be con-cluded that addition of the co-polymer to PES membranes could im-prove the cytocompatibility, and the modified membranes have thepotential to be used as bioartificial liver supports.
4. Conclusion
A good approach to modify polymeric membranes with excel-lent biocompatibility has been provided by blending them with a

3380 F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381
novel amphiphilic triblock co-polymer PVP-b-PMMA-b-PVP, whichwas synthesized via RAFT polymerization. The block co-polymercan be directly blended with PES to prepare PES flat sheet and hol-low fiber membranes with a PVP block brush layer on their surface.The blended membranes have better blood compatibility (BSAadsorption, platelet adhesion, and blood coagulation time) com-pared with the PES membrane due to modification on blendingwith the amphiliphilic block co-polymer. The modified membranesalso showed good ultrafiltration and protein anti-fouling proper-ties. Furthermore, the cytocompatibility of the modified mem-branes was improved. These results indicate that the modifiedmembranes have the potential to be used in blood purification,including hemodialysis and bioartificial liver support, and thestudy has provided useful information for practical application ofthe membranes.
Acknowledgements
This work was financially sponsored by the National NaturalScience Foundation of China (grants nos. 50973070, 51073105and 30900691) and Sichuan Youth Science and Technology Foun-dation (08ZQ026-038). We would also like to thank our laboratorymembers for their generous help, and gratefully acknowledge thehelp of Ms H. Wang, of the Analytical and Testing Center at SichuanUniversity, for the SEM, and Ms Liang, of the Department ofNephrology at West China Hospital, for the human fresh bloodcollection.
Appendix A. Figures with essential colour discrimination
Certain figures in this article, particularly Figure 4, is difficult tointerpret in black and white. The full colour image can be found inthe on-line version, at doi:10.1016/j.actbio.2011.05.026.
References
[1] Samtleben W, Dengler C, Reinhardt B, Nothdurft A, Lemke HD. Comparison ofthe new polyethersulfone high-flux membrane DIAPES HF800 withconventional high-flux membranes during on-line haemodiafiltration.Nephrol Dial Transplant 2003;18(11):2382–6.
[2] Huang XJ, Guduru D, Xu ZK, Vienken J, Groth T. Blood compatibility andpermeability of heparin-modified polysulfone as potential membrane forsimultaneous hemodialysis and LDL removal. Macromol Biosci2011;11(1):131–40.
[3] Wang T, Wang YQ, Su YL, Jiang ZY. Antifouling ultrafiltration membranecomposed of polyethersulfone and sulfobetaine copolymer. J Membr Sci2006;280(1/2):343–50.
[4] Lin YC, Brayfield CA, Gerlach JC, Rubin JP, Marra KG. Peptide modification ofpolyethersulfone surfaces to improve adipose-derived stem cell adhension.Acta Biomater 2009;5(5):1416–24.
[5] Peppas NA, Langer R. New challenges in biomaterials. Science1994;263(5154):1715–20.
[6] Ishihara K, Fukumoto K, Iwasaki Y, Nakabayashi N. Modification of polysulfonewith phospholipids polymer for improvement of the blood compatibility. Part1: Surface characterization. Biomaterials 1999;20(17):1545–51.
[7] Ishihara K, Fukumoto K, Iwasaki Y, Nakabayashi N. Modification of polysulfonewith phospholipid polymer for improvement of the blood compatibility. Part2: Protein adsorption and platelet adhesion. Biomaterials 1999;20(17):1553–9.
[8] Takashi H, Yasuhiko I, Kazuhiko I. Preparation and performance of protein-adsorption-resistant asymmetric membrane composed of polysulfone/phospholipid polymer blend. Biomaterials 2001;22(3):243–51.
[9] Zhu LP, Yi Z, Liu F, Wei XZ, Zhu BK, Xu YY. Amphiphilic graft copolymers basedon ultrahigh molecular weight poly(styrene-alt-maleic anhydride) withpoly(ethylene glycol) side chains for surface modification ofpolyethersulfone membranes. Eur Polym J 2008;44(6):1907–14.
[10] Ulbricht M, Matuschewski H, Oechel A, Hicke HG. Photo-induced graftpolymerization surface modifications for the preparation of hydrophilic andlow-proten-adsorbing ultrafitration membranes. J Membr Sci 1996;115(1):31–47.
[11] Susanto H, Ulbricht M. Photografted thin polymer hydrogel layers on PESultrafiltration membranes: characterization, stability, and influence onseparation performance. Langmuir 2007;23(14):7818–30.
[12] Dattatray S, Ellen W, Fisher R. Hydrophilic modification of polyethersulfonemembranes by low temperature plasma-induced graft polymerization. JMembr Sci 2002;209(1):255–69.
[13] Bernacca GM, Gulbransen MJ, Wilkinson R, Wheatley DJ. In vitro bloodcompatibility of surface-modified polyurethanes. Biomaterials1998;19(13):1151–65.
[14] Yuan J, Zhang J, Zang XP, Shen J, Lin S. Improvement of blood compatibility oncellulose membrane surface by grafting betaines. Colloid Surfaces B Biointerf2003;30(1/2):147–55.
[15] Su BH, Fu P, Li Q, Tao Y, Li Z, Zao HS, et al. Evaluation of polyethersulfonehighflux hemodialysis membrane in vitro and in vivo. J Mater Sci Mater Med2008;19(2):745–51.
[16] Wang YQ, Wang T, Su YL, Peng FB, Wu H, Jiang ZY. Protein-adsorption-resistance and permeation property of polyethersulfone and soybeanphosphatidylcholine blend ultrafiltration membranes. J Membr Sci2006;270(1/2):108–14.
[17] Liu ZB, Deng XP, Zhao CS. BSA hybrid synthesized polymer. Chin Chem Lett2006;17(11):1519–22.
[18] Liu ZB, Deng XP, Wang M, Chen JX, Zhang AM, Gu ZW, et al. BSA-modifiedpolyethersulfone membrane: preparation, characterization andbiocompatibility. J Biomat Sci 2009;20(3):377–97.
[19] Wang HT, Yu T, Zhao CY, Du QY. Improvement of hydrophilicity and bloodcompatibility on polyethersulfone membrane by adding polyvinylpyrrolidone.Fiber Polym 2009;10(1):1–5.
[20] Henry M, Bertrand P. Surface composition of insulin and albumin adsorbed onpolymer substrates as revealed by multivariate analysis of ToF-SIMS data. SurfInterface Anal 2009;41(3):105–13.
[21] Zhang LF, Liang Y, Meng LZ, Wang C. Characterization of complexation of PVPcopolymer with DNA. Polym Adv Technol 2009;20(4):410–5.
[22] Haaf F, Sanner A, Straub F. Polymers of N-vinylpyrrolidone: synthesis,characterization and uses. Polym J 1985;17(1):143–52.
[23] George KA, Wentrup-Byrne E, Hill DJT, Whittaker AK. Investigation into thediffusion of water into HEMA-co-MOEP hydrogels. Biomacromolecules2004;5(4):1194–9.
[24] Barzin J, Feng C, Khulbe KC, Matsuura T, Madaeni SS, Mirzadeh H.Characterization of polyethersulfone hemodialysis membrane byultrafiltration and atomic force microscopy. J Membr Sci 2004;237(1/2):77–85.
[25] Mosqueda-Jimenez DB, Narbaitz RM, Matsuura T. Effects of preparationconditions on the surface modification and performance of polyethersulfoneultrafiltration membranes. J Appl Polym Sci 2006;99(6):2978–88.
[26] Wan LS, Xu ZK, Wang ZG. Leaching of PVP from polyacrylonitrile/PVP blendingmembranes: a comparative study of asymmetric and dense membranes. JPolym Sci Part B Polym Phys 2006;44(10):1490–8.
[27] Sun MP, Su YL, Mu CX, Jiang ZY. Improved antifouling property of PESultrafiltration membranes using additive of silica-PVP nanocomposite. Ind EngChem Res 2010;49(2):790–6.
[28] Li LL, Yin ZH, Li FL, Xiang T, Chen Y, Zhao CS. Preparation and characterizationof poly(acrylonitrile-acrylic acid-N-vinyl pyrrolidinone) terpolymer blendedpolyethersulfone membranes. J Membr Sci 2010;349(1/2):56–64.
[29] Zou Wen, Huang Y, Luo J, Bai PL, Zhao CS. Modification of polyethersulfonemembrane by grafting bovine serum albumin on the surface ofpolyethersulfone/poly(acrylonitrile-co-acrylic acid) blended membrane. JMembr Sci 2009;329(1/2):46–55.
[30] Cai XJ, Chen S, Chen L. Solvent-free free-radical frontal polymerization: a newapproach to quickly synthesize poly(N-vinylpyrrolidone). J Polym Sci Part APolym Chem 2008;45(6):2117–85.
[31] Uyanik N. Synthesis and characterization of five-block copolymers prepared byvinyl pyrrolidinone and a macro initiator with poly(dimethylsiloxane) andpolycaprolactone. J Appl Polym Sci 1997;64(10):1961–9.
[32] Bilalis P, Pitsilalis M, Hadjichristidis N. Controlled nitroxide-mediated andreversible addition–fragmentation chain transfer polymerization of N-vinylpyrrolidone: synthesis of block copolymers with styrene and 2-vinylpyridine. J Polym Sci Part A Polym Chem 2006;44(1):659–65.
[33] Bilalis P, Zorba G, Pitsilalis M, Hadjichristidis N. Synthesis of poly(n-hexylisocyanate-b-N-vinylpyrrolidone) block copolymers by the combinationof anionic and nitroxidemediated radical polymerizations: micellizationproperties in aqueous solutions. J Polym Sci Part A Polym Chem 2006;44(19):5719–28.
[34] Ray B, Kotani M, Yamago S. Highly controlled synthesis of poly(N-vinylpyrrolidone) and its block copolymers by organostibine-mediated livingradical polymerization. Macromolecules 2006;39(16):5259–65.
[35] Li YY, Cheng H, Zhu JL, Yuan L, Dai Y, et al. Temperature- and pH-sensitivemulticolored micellar coplexes. Adv Mater 2009;21(23):2402–6.
[36] Li YY, Dai Y, Zhang XZ, Zhuo RX. The tuned-morphology studies of thecomplexes between poly(N-isopropylacrylamide)-b-poly(vinylpyridine) andpoly(N-isopropylacrylamide-co-hydroxylethyl methacrylate)-b-poly(vinyl-phenol). J Colloid Interf Sci 2008;328(1):211–5.
[37] Lu XJ, Gong SL, Meng LZ, Li C, Yang S, Zhang LF. Controllable synthesis ofpoly(N-vinylpyrrolidone) and its block copolymers by atom transfer radicalpolymerization. Polymer 2007;48(10):2835–42.
[38] Yao X, Yao HW, Li GY, Li YT. Biomimetic synthesis of needle-like nano-hydroxyapatite templated by double-hydrophilic block copolymer. J Mater Sci2010;45(7):1930–6.
[39] Debuigne A, Willet N, Jerome R, Detrembleur C. Amphiphilic poly(vinylacetate)-b-poly(N-vinylpyrrolidone) and novel double hydrophilic poly(vinyl

F. Ran et al. / Acta Biomaterialia 7 (2011) 3370–3381 3381
alcohol)-b-poly(N-vinylpyrrolidone) block copolymers prepared by cobalt-mediated radical polymerization. Macromolecules 2007;40(20):7111–8.
[40] Bartolozzi I, Solaro R, Schacht E, Chiellini E. Hydroxyl end-capped macromersof N-vinyl-2-pyrrolidinone as precursors of amphiphilic block copolymers. EurPolym J 2007;43(11):4628–38.
[41] Hussain H, Tan BH, Gudipati CS, Liu Y, He CB, Davis TP. Synthesis and self-assembly of poly(styrene)-b-poly(N-vinylpyrrolidone) amphiphilic diblockcopolymers made via a combined ATRP and MADIX approach. J Polym SciPart A Polym Chem 2008;46(16):5604–15.
[42] Hussain H, Tan BH, Gudipati CS, He CB, Liu Y, Davis TP. Micelle formation ofamphiphilic polystyrene-b-poly(N-vinylpyrrolidone) diblock copolymer inmethanol and water-methanol binary mixtures. Langmuir 2009;25(10):5557–64.
[43] Luo YL, Wang AR, Yuan JF, Gao QY. Preparation, characterization and drugrelease behavior of polyion complex micelles. Intern J Pharm 2009;374(1/2):139–44.
[44] Luo YL, Yao XJ, Yuan JF, Ding T, Gao QY. Preparation and drug controlled-release of polyion complex micelles as drug delivery systems. Colloid Surf BBiointerf 2009;68(2):218–24.
[45] Benahmed A, Ranger M, Leroux JC. Novel polymeric micelles based on theamphiphilic diblock copolymer poly(N-vinyl-2-pyrrolidone)-b-poly(D, L-lactide). Pharma Res 2001;18(3):323–8.
[46] Hadjichristidisa N, Iatroua H, Pitsikalisa M, Pispasb S, Avgeropoulosc A. Linearand non-linear triblock terpolymers. Synthesis, self-assembly in selectivesolvents and in bulk. Prog Polym Sci 2005(7):725–82.
[47] Matyjaszewski K, Müller AHE. 50 years of living polymerization. Prog PolymSci 2006;31(12):1039–40.
[48] Braunecker WA, Matyjaszewski K. Controlled/living radical polymerization:features, developments, and perspectives. Prog Polym Sci 2007;32(1):93–146.
[49] Qiu J, Matyjaszewski K. Controlled/living radical polymerization in aqeousmedia. Prog Polym Sci 2001;26(10):2083–134.
[50] John TL, Debby F, Ronald S. Functional polymers from novel carboxyl-terminated trithiocarbonates as highly efficient RAFT agents.Macromolecules 2002;35(18):6754–6.
[51] Yang Q, Chung TS, Weber M. Microscopic behavior of polyvinylpyrrolidonehydrophilizing agents on phase inversion polyethersulfone hollow fibermembranes for hemofiltration. J Membr Sci 2009;326(2):322–31.
[52] Qian B, Li J, Wei Q, Bai PL, Fang BH, Zhao CS. Preparation and characterizationof pH-sensitive polyethersulfone hollow fiber membrane for flux control. JMembr Sci 2009;344(1/2):297–303.
[53] Moad G, Rizzardo E, Thang SH. Radical addition–fragmentation chemistry inpolymer synthesis. Polymer 2008;49(5):1079–131.
[54] Nie SQ, Ran F, He C, Zhao PF, Wei XH, Li J, et al. Synthesis of amphiphilic tri-block copolymer poly(vinyl pyrrolidone)-b-poly(methyl methacrylate)-b-poly(vinyl pyrrolidone) for the modification of polyethersulfone membrane.Chin Chem Lett 2010;22(3):370–3.
[55] Masahide T, Georges B. Low protein fouling synthetic membranes by UVassisted-surface grafting modification: varying monomer type. J Membr Sci2004;231(1/2):147–57.
[56] Masahide T, James EK, Belfort G. Low fouling synthetic membranes by UV-assisted graft polymerization: monomer selection to mitigate fouling bynatural organic matter. J Membr Sci 2003;222(1/2):59–70.
[57] Jia ZF, Xu XW, Fu Q, Huang JL. Synthesis and self-assembly morphologies ofamphiphilic multiblock copolymers [poly(ethylene oxide)-b-polystyrene]n viatrithiocarbonate-embedded PEO macro-RAFT Agent. J Polym Sci Part A PolymChem 2006;44(20):6071–82.
[58] Zhang GZ, Liu L, Zhao Y, Ning FL, Jiang M, Wu Q. Self-assembly of carboxylatedpoly(styrene-b-ethylene-co-butylene-b-styrene) triblock copolymer chains inwater via a microphase inversion. Macromolecules 2000;33(17):6340–3.
[59] Kuang M, Duan HW, Wang J, Jiang M. Structural factors of rigid-coil polymerpairs influencing their self-assembly in common solvent. J Phys Chem B2004;108(41):16023–9.
[60] Duan HW, Chen DY, Jiang M, Gan WJ, Li SJ, Wang M, et al. Self-assembly ofunlike homopolymers into hollow spheres in nonselective solvent. J Am ChemSoc 2001;123(48):12097–8.
[61] Liu J, Liu T, Statish K. Effect of solvent solubility parameter on SWNT dipersionin PMMA. Polymer 2005;46(10):3419–24.
[62] Brandrup J, Immergut EH, Gurlke EA. Polymer Handbook, IVth edn. NewYork: John Wiley & Sons; 1999.
[63] Lafreniere L, Talbot FDF, Matsuura T, Sourirajan S. Effect of PVP additive on theperformance of PES ultrafiltrationmembrane. Ind Eng Chem Res 1987;26:2385–9.
[64] Miyano T, Matsuura T, Sourirajan S. Effect of polyvinylpyrrolidone additive onthe pore-size and the pore-size distribution of polyethersulfone (Victrex)membranes. Chem Eng Commun 1993;119:23–39.
[65] Martinelli E, Menghetti S, Galli G, Glisenti A, Krishnan S, Paik MY, et al. Surfaceengineering of styrene/PEGylated-fuoroalkyl styrene block copolymer thinfilms. J Polym Sci Part A Polym Chem 2009;47:267–84.
[66] Krishnan S, Paik MY, Ober CK, Martinelli E, Galli G, Sohn KE, et al. NEXAFSdepth profilling of surface segregation in block copolymer thin films.Macromolecules 2010;43(10):4733–43.
[67] Neto C, James M, Telford AM. On the composition of the top layer ofmicrophase separated thin PS-PEO films. Macromolecules 2009;42(13):4801–8.
[68] Krishnan S, Ward RJ, Hexemer A, Sohn KE, Lee KL, Angert ER. Surfaces offluorinated pyridinium block copolymers with enhanced antibacterial activity.Langmuir 2006;22(26):11255–66.
[69] Krishnan S, Wang N, Ober CK, Finlay JA, Callow ME, Callow JA.Comparison of the fouling release properties of hydrophobic fluorinatedand hydrophilic PEGylated block copolymer surfaces: attachment strengthof the diatom navicula and the green alga ulwa. Biomacromolecules2006;7(5):1449–62.
[70] Krishnan S, Ayothi R, Hexemer A, Finlay JA, Sohn KE, Perry R, et al. Anti-biofouling properties of comblike block copolymers with amphiphilic sidechains. Langmuir 2006;22(11):5075–86.
[71] Nishimura T. Polymer materials for blood purification. In: Tsuruta T, Hyashi T,Kataoka K, Ishihara K, Kimura Y, editors. Biomedical application of polymericmaterials. Boca Raton, FL: CRC Press; 1993. p. 191–218.
[72] Jane YP, Metin HA, Ariya A, Kuhlman W, Mayes AM. Polysulfone-graft-poly(ethylene glycol) graft copolymers for surface modification of polysulfonemembranes. Biomaterials 2006;27(6):856–65.
[73] Kim YJ, Kang IK, Huh MW, Yoon SC. Surface characterization and in vitroblood compatibility of poly(ethylene terephthalate) immobilized with insulinand/or heparin using plasma glow discharge. Biomaterials 2000;21(2):121–30.
[74] Guo Z, Meng S, Zhong W, Du QG, Chou LL. Self-assembly of silanatedpoly(ethylene glycol) on silicon and glass surfaces for improvedhomocompatibility. Appl Surf Sci 2009;255(15):6771–80.
[75] Lin WC, Liu TY, Yang MC. Hemocompatibility of polyacrylonitrile dialysismembrane immobilized with chitosan and heparin conjugate. Biomaterials2004;25(10):1947–57.
[76] Winchester JF, Audia PF. Extracorporeal strategies for the removal of middlemolecules. Semin Dialysis 2006;19(2):110–4.
[77] Bjorkman PJ, Burmeister WP. Structures of two classes of MHC moleculeselucidated: crucial differences and similarities. Curr Opin Struct Biol1994;4(6):852–6.
[78] Strominger JL. Human histocompatibility proteins. Immunol Rev 2002;185(1):69–77.
[79] PRahimpour A, Madaeni SS, Zereshki S, Mansourpanah Y. Preparation andcharacterization of modified nano-porous PVDF membrane with highantifouling property using UV photo-grafting. Appl Surf Sci 2009;255(16):7455–61.
[80] Serkiz SM, Perdue EM. Isolation of dissolved organic matter from theSuwannee river using reverse osmosis. Water Res 1990;24(12):1471–7.
[81] Zhao WF, He C, Wang HY, Su BH, Sun SD, Zhao CS. Improved antifoulingproperty of polyethersulfone hollow fiber membranes using additive ofpoly(ethylene glycol) methyl ether-b-poly(styrene) copolymers. Ind EngChem Res 2011;50(6):3295–303.
[82] Naruse K, Tang W, Makuuchi M. Artificial and bioartificial liver support: areview of perfusion treatment for hepatic failure patients. World JGastroenterol 2007;13(10):1516–21.
[83] Gautier A, Ould-Dris A, Dufresne M, Paullier P, Harten BV, Lemke HD, et al.Hollow fiber bioartificial liver: physical and biological characterization withC3A cells. J Membr Sci 2009;341(1/2):203–13.
[84] Rajaraman R, Rounds DE, Yen SPS, Rembaum A. A scanning electronmicroscope study of cell adhesion and spreading in vitro. Exp Cell Res1974;88(2):327–39.
[85] Sakai Y, Nakazawa K. Technique for the control of spheroid diameter usingmicrofabricated chips. Acta Biomater 2007;3(6):1033–40.
[86] Ijima H, Nakazawa K, Mizumoto H, Matsushita T, Funatsu K. Formation of aspherical multicellular aggregate (spheroid) of animal cells in the pores ofpolyurethane foam as a cell culture substratum and its application to a hybridartificial liver. J Biomater Sci 1998;9(7):765–78.





























