Embed Size (px)
Citation preview

1
Marketing Authorization Procedures and Quality Marketing Authorization Procedures and Quality
Management for Generics in TaiwanManagement for Generics in Taiwan
Meir-Chyun Tzou, Ph.D.
Director, Division of Drugs and New Biotechnology Products,
Taiwan Food and Drug Administration,
Department of Health,
Taiwan, R.O.C.2011.06.03
Generic Medicines in the Legal Systems of the Republic of China and the Federal Republic of Germany

2
OutlineOutline
Organization and Responsibility
Management of Drug Quality-product Life Cycle ManagementPre-Marketing ApprovalPost-Marketing Management
Issues of Intellectual Property Right for Drug Approval
Future perspectives

3333
Taiwan FDA (TFDA) was inaugurated on Jan. 1, 2010
TFDA supersedes the following 4 bureaus of Department of Health Bureau of Food SafetyBureau of Pharmaceutical AffairsBureau of Food and Drug AnalysisBureau of Controlled Drugs
Establishment of Taiwan FDAEstablishment of Taiwan FDA
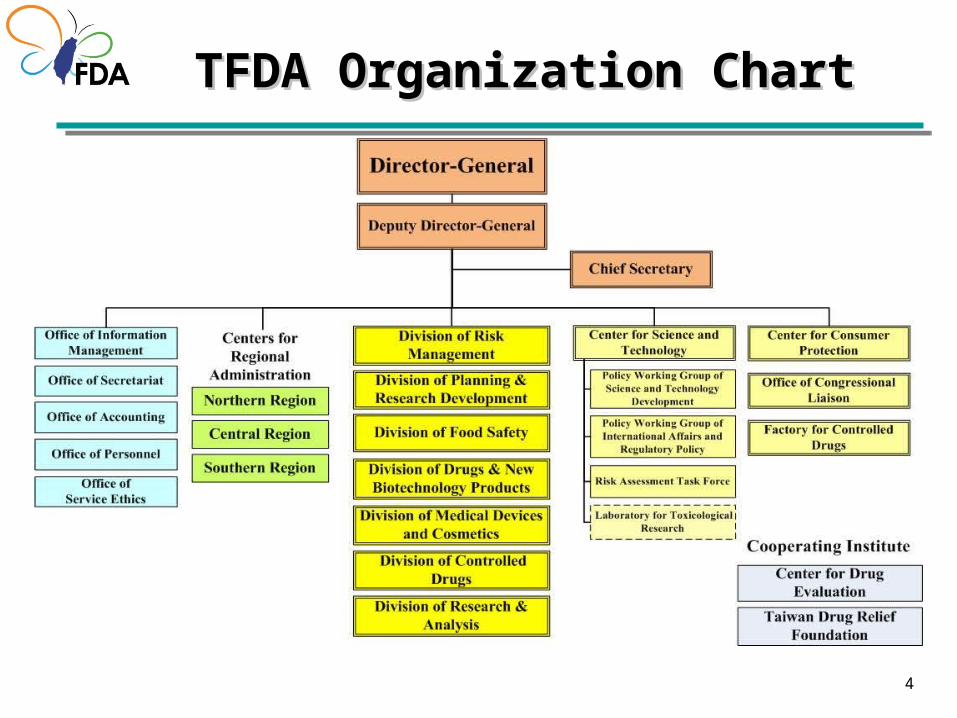
4444
TFDA Organization ChartTFDA Organization Chart

5
Core Value of TFDACore Value of TFDA
From Product Centered to Consumer Centered
Risk Analysis and risk communication
Drug Management
Food Management
Consumer Protection

6
學名藥22875
84.13%原料藥23188.53%
罕見疾病藥品35 件
新藥1649件
生物藥品309 件1.14%
Statistics on Pharmaceutical LicensesStatistics on Pharmaceutical Licenses(2010)(2010)
New drugGeneric drug
API
Biologics
Orphan Drugs
22857 84.13%

777
Statistics on Pharmaceutical LicensesStatistics on Pharmaceutical Licenses (2010) (2010)
Domestic Import TotalRx drug 13449 3239 16688
Non-Rx drug 7543 619 8162
Total 20992 3858 24850
Rx: Non-RX 1.8:1 5.2:1 2:1
Domestic (%)
Import (%) Total (%)
New Drug 493 (30) 1156 (70) 1649 (100)
Generic Drug 20499 (90) 2358 (10) 22857 (100)
API 560 (24) 1758 (76) 2318 (100)
Total no. of Licenses 21552 (79) 5616 (21) 27168 (100)
ratio ~1:42:1.2 ~1:2.3:1.5 ~1:14:1.4
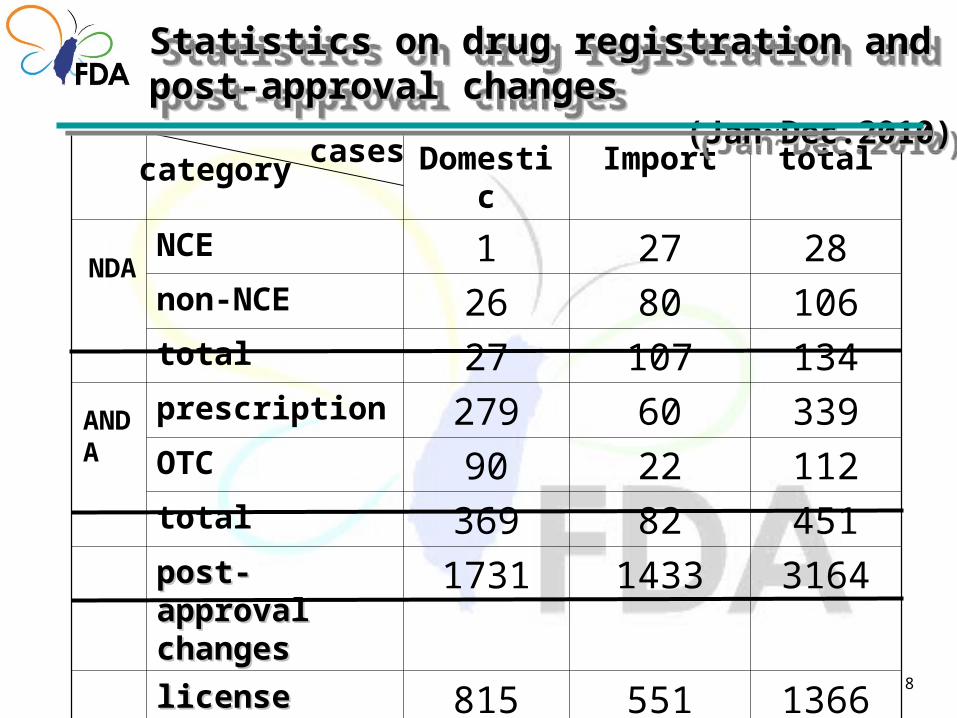
8
Domestic Import total
NCE 1 27 28non-NCE 26 80 106total 27 107 134prescription 279 60 339OTC 90 22 112total 369 82 451post-approval post-approval changeschanges
1731 1433 3164
license license extensionsextensions
815 551 1366
casescategory
NDA
ANDA
Statistics on drug registration and post-approval Statistics on drug registration and post-approval changes changes (Jan~Dec.2010)(Jan~Dec.2010)
Statistics on drug registration and post-approval Statistics on drug registration and post-approval changes changes (Jan~Dec.2010)(Jan~Dec.2010)

9
Management of Drug Quality-Management of Drug Quality-product Life Cycle Managementproduct Life Cycle Management

Management of Drug Quality-product Life Management of Drug Quality-product Life Cycle ManagementCycle Management
Basic research
Pre- clinical
ClinicalTrials
Product Launch
ProductionMarketing
& Sales
Review
Inspection
CTD : Safety 、 Efficacy 、 Quality
GMP/GTP
GLP 、 GCP GPvP
Analysis Testing / Trial/Analysis
(審查 )
(稽查 )
(檢驗 )10

1111
1980 1990 2000 2010
Milestones on Drug RegulationMilestones on Drug Regulation
1982GMP
1982GMP
1983BA/BE
1983BA/BE
1999cGMP
1999cGMP
2009DMF
2009DMF
2010PIC/S GMP
2010PIC/S GMP
1993Local
clinical trial
1993Local
clinical trial1996 GCP
1996 GCP
1998GLP
1998GLP
*Bridging Study Evaluation in accordance with ICH E5*Bridging Study Evaluation in accordance with ICH E5
2000BS *
Evaluation /ICH E5
2000BS *
Evaluation /ICH E5
2001Pivotal trial/
early phase trial
2001Pivotal trial/
early phase trial
1998CDE
1998CDE
2001TDRF
2001TDRF
2010TFDA
2010TFDA
1983PV/PMS
1983PV/PMS
1998ADR
1998ADR
2008GPvP
2008GPvP

121212
Milestones on Drug RegulationMilestones on Drug Regulation1982 Good Manufacturing Practice (GMP)1987 Bioavailability / Bioequivalence Guideline (BA/BE)1993 Safety monitoring system-the requirement of local clinical trial1996 Good Clinical Practice (GCP) 1998 Good Laboratory Practices (GLP)1999 Establishment of Adverse Drug Reaction reporting system 1999 Amendment of current GMP (cGMP) 2004 Implementation of bridging study evaluation (BSE) 2005 Revision of Pharmaceutical Affairs Act (Data Exclusivity & compulsory requirement of serious ADR reporting)2008 Good Pharmacovigilance Practice (GPvP)2008 Guideline for Biosimilar Product Review and Approval2009 Drug Master File (DMF)2010 Implementation of PIC/S GMP2010 Amendment of Guideline for Biosimilar Product Review and Approval
2010 Develop strategies for review guidelines of tNCE and Botanical New Drug (in process)
2010 Relaxation of CPP requirement (draft)
12

13
Pre-Marketing ApprovalPre-Marketing Approval

14
Pre-marketing reviewPre-marketing review
New Drug Generic drug
Safety
Efficacy
Pharm / Tox
PK/PD/BA/BE
Clinical trials
Bioequivalence (BE) as a surrogate to clinical trial
Quality Chemistry, Manufacturing and Controls, CMC Including Active Pharmaceutical Ingredient (API) ,
excipients, process control, validation, stability, etc.
GLP, GCP, cGMP Labeling ( direction of use)

15
Regulatory requirement on BE studiesRegulatory requirement on BE studies
Regulations Guideline of Bioavailability and Bioequivalence
(BA/BE) studies (1987) Regulations of BA/BE Studies (2009)
Drug ApplicationNew drugs Generic drugs
—Since 1983—Retrospectively request BE studies for drugs approved before
1983 with BE concerne.g., Diltiazem 、 Glyburide 、 Furosemide 、 Isosorbide
dinitrate 、 Atenolol 、 Nifedipine 、 Rifampin 、 Digoxin 、 Carbamazepine

BA/BEBA/BE studies inspectionstudies inspection
Implement inspection from 2002Clinical phase should follow GCPLaboratory phase should follow GLPOther Regulations of BA/BE Studies Reviewer initiated inspection
16

17
PIC/S GMP
1982◆
1999◆
cGMPGMP
◆
2010.11◆
1988◆
2005
211 230163
165
550
0
100
200
300
400
500
600
21 pharmaceutical manufacturers are in
compliance with PIC/S GMP
Milestones of Pharmaceutical GMP Development Milestones of Pharmaceutical GMP Development in Taiwanin Taiwan
number of domestic pharmaceutical manufacturer
Overseas Inspection since 2002

Regulatory requirement on Active Regulatory requirement on Active Pharmaceutical Ingredients (API)Pharmaceutical Ingredients (API)
DMF for API of NDA and biological products
2002 GMP Guide for Active Pharmaceutical Ingredients
2009 Announced DMF technical information review form and application notes for API.
18

19
Post-marketing ManagementPost-marketing Management

20
Regulation on post-approval changesRegulation on post-approval changes
Types of Post-approval Changes Scale of manufacturing, Manufacturing process,
equipment, site, manufacturer, etc.Particle size, crystalline form, polymorphs, in-process,
controls, product release specification, etc.Synthetic procedures, source of API and excipients,
supplier, etc.Regulation Requirement
Scale-Up and Post Approval Changes (SUPAC) ( 2001 public Announcement )
For products that have passed the BE testing and registered for marketing, any changes, depending on the level and extent of change are required to submit Bioequivalence testing report or Dissolution Rate Profile to assure and verify its quality.

21
Post-approval commitmentPost-approval commitment
Surveillance on Safety, Efficacy and Quality Post-marketing Surveillance, Phase-IV trialADR/quality defect reporting and investigationREMS/RMP
Maintenance for Drug Quality-Life Cycle Management (industry’s role)Well controlled process and quality system
— batch to batch releaseOn-going stability protocolPost-approval changes, annual report Inspection

22
The role of Government and IndustryThe role of Government and Industry in ensuring in ensuring Drug qualityDrug quality
Government’s role Industry’s role
Review Review to ensure S 、 E&Q Provide data for S 、 E&Qnon-clinical, clinical trial, CMC
Testing Batch release for biologics
No testing for other drugs (with exceptions)
In process control and batch to batch release for drugs and biologics
Inspection Inspection to assure compliance
Comply with GXP-
to assure data integrity and honest communication
It is mainly the industry’s responsibility, not the government’s,to ensure product quality throughout product life cycle

232323
Post-marketing Post-marketing ManagementManagement System System
23
Post-marketing Management systemPost-marketing Management system
Post-approval changes
Post-approval changes
Post-approvalcommitment
Post-approvalcommitment
ADR reporting system
ADR reporting system
Product quality defect reporting
system
Product quality defect reporting
system
Compliance Compliance
GMPGMPSafety
PharmacovigilanceSafety
Pharmacovigilance
ReviewReview ReviewReviewReview/Testing
Review/Testing
InspectionInspection
Quality surveillance
Quality surveillance

24
Post-marketing product quality
and safety surveillance
Post-marketing product quality
and safety surveillance
Quality surveillance
Quality surveillance
Safety surveillance
Safety surveillance
Drug Product quality defect reporting system
Drug Product quality defect reporting system
ActiveActive
ActiveActive
National Quality surveillance Program
National Quality surveillance Program
National ADR reporting system
National ADR reporting system
Manufacturer : Drug safety report on a regular
basis(PSUR)
Manufacturer : Drug safety report on a regular
basis(PSUR)
Government : ADR active Monitoring Network
Government : ADR active Monitoring Network
Reassessment / InspectionLabeling change
Withdrawal/Recall
Reassessment / InspectionLabeling change
Withdrawal/Recall
SurveillanceSurveillance
Post-marketing Safety and Quality Post-marketing Safety and Quality Surveillance-Risk ManagementSurveillance-Risk Management
PassivePassive
PassivePassive
Therapeutic Inequivalence Reporting System
Therapeutic Inequivalence Reporting System
24

Issues of Intellectual Property Right Issues of Intellectual Property Right for Drug Approvalfor Drug Approval
25

26
Issues of Patent
Issues of Exclusivity
Issues of Research ExemptionIssues ofIntellectual
Property Right for Drug Approval
Issues of Copyright

Issues of PatentIssues of Patent
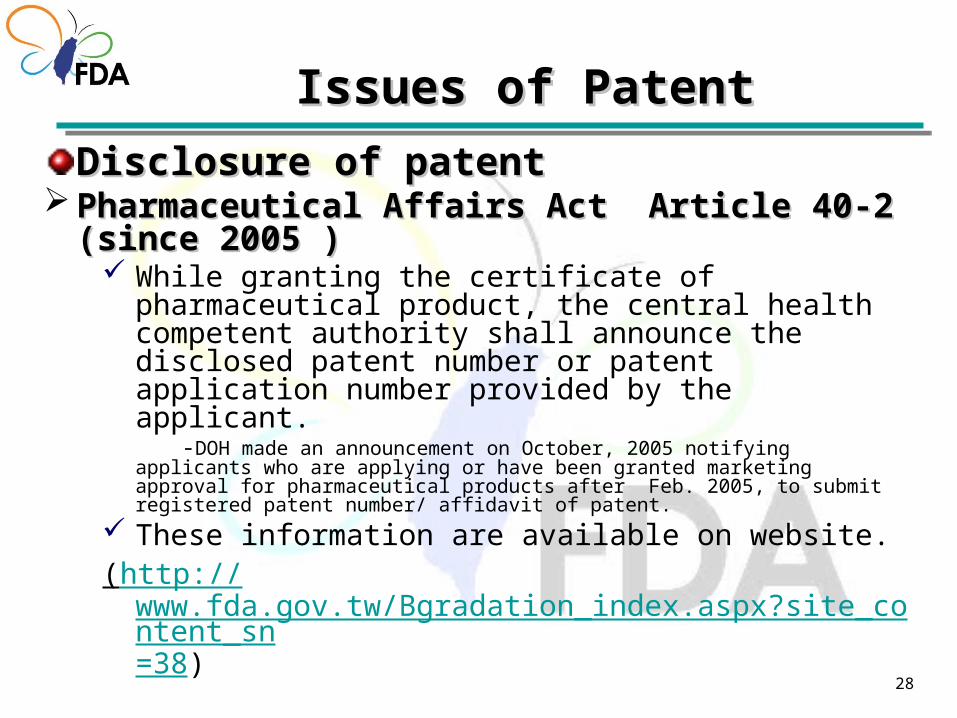
Disclosure of patent
Declaration of patent non-infringement
The review of generic drug and the information of patents are normally linked
Review of patent infringement cases- Intellectual Property Court
27
28
Issues of PatentIssues of Patent
Disclosure of patentDisclosure of patent Pharmaceutical Affairs Act Article 40-2 (since 2005 Pharmaceutical Affairs Act Article 40-2 (since 2005
)) While granting the certificate of pharmaceutical product,
the central health competent authority shall announce the disclosed patent number or patent application number provided by the applicant.
-DOH made an announcement on October, 2005 notifying applicants who are applying or have been granted marketing approval for pharmaceutical products after Feb. 2005, to submit registered patent number/ affidavit of patent.
These information are available on website.(http://
www.fda.gov.tw/Bgradation_index.aspx?site_content_sn=38)

29
Issues of PatentIssues of Patent
Declaration of patent non-infringement Review Regulations for Registration and Market
Approval of Pharmaceuticals Article 19Applicants should submit an affidavit to declare that the
applicant has full legal responsibility if the applied product infringe the patent of others.
The review of generic drug and the information of patents are normally linked.

Issues of PatentIssues of Patent
Review of patent infringement cases- Intellectual Property Court According to Taiwan’s Patent Act, within valid patent
protection period, generic drug cannot enter the market or be sold.
Patent infringement will be granted by the patentee if the generic drug applicants violate the rule.
Patent-related litigation and the quality of review had been enhanced and accelerated since the Intellectual Property Court was established on July 1st, 2008.
30

31
Issues of Issues of Research Exemption Exemption
Exemption for Research useExemption for Research use Pharmaceutical Affairs Act Article 40-2 Paragraph 5Pharmaceutical Affairs Act Article 40-2 Paragraph 5
The patent right of the new drug shall not be applicable to researches, teachings, or testing prior to the application for registration by the pharmaceutical firms.
Patent Act Article 60(Amending)

32
Issues of Exclusivity Issues of Exclusivity
Historical exclusivity analogyHistorical exclusivity analogy 1993 Safety monitoring system-the requirement of local
clinical trial for NDA (Taiwan-US Intellectual Property Right Agreement) Pipeline protection of new drug before the Patent Act was amended
in 1986.Barrier for generic drug applicationWithin five years after the issuance of a license for new drug , the
second applicant is required to submit the same scale of clinical trial data as the first applicant.
1997 New indication2004 Implementation of bridging study evaluation (BSE)

33
Issues of Data exclusivityIssues of Data exclusivity
Within five years after the issuance of a license for new drug of new chemical entity, any other pharmaceutical firm may not apply for evaluation and registration of the same items by citing the data submitted by the licensee without such licensee’s authorization.
After three years of the issuance of a license for new drug of new chemical entity, other pharmaceutical firm may apply for registration of drugs of the same substance, the same dosage form, the same dose, and the same dose unit according to this Act and related laws or regulations; the drug license may be issued on the next day to the expiration of five years after the issuance of license to such new drug of new chemical entity.
Pharmaceutical Affairs Act Article 40-2Pharmaceutical Affairs Act Article 40-2

34
Issues of Data exclusivityIssues of Data exclusivity
Pharmaceutical Affairs Act Article 40-2Pharmaceutical Affairs Act Article 40-2The second paragraph hereof can only be applicable
with the compliance that application for registration of a new drug of new chemical entity shall be made to the Central Competent Health Authority within three years after it is first approved for marketing in any country.

35
Generic drug labeling Review Regulations for Registration and Market
Approval of Pharmaceuticals Article 20Generic drug labeling of the surveillance drugs*, should
follow the first approved labeling; labeling of the non-surveillance drugs** should be translated according to the innovator’s labeling.
But drug labeling has not been excluded from the copyright act, which might cause the lawsuit.
*surveillance drugs: chemical entity first approved after 1983.
**non-surveillance drugs : chemical entity first approved before 1983.
Issues of Copyright Issues of Copyright

36
Drug labeling – fair use for public benefitDrug labeling – fair use for public benefit Copyright Act Article 65Copyright Act Article 65
Fair use of a work shall not constitute infringement on economic rights in the work. In determining whether the exploitation of a work complies with the provisions of Articles
44 through 63, or other conditions of fair use, all circumstances shall be taken into account, and in particular the following facts shall be noted as the basis for determination.
The court decision for lawsuit cases : Drug labeling should refer to Copyright Act Article 52 “Within a reasonable scope, it may be used for necessary or other legitimate purposes”Drug labeling excluding from copyright
TFDA is planning to amend the Pharmaceutical Affairs Act Article 48-2Pharmaceutical Affairs Act Article 48-2, to stipulates the suitability of excluding drug labeling from Copyright Act.
Issues of Copyright Issues of Copyright

Future perspectivesFuture perspectives of Intellectual of Intellectual Property Right for Drug ApprovalProperty Right for Drug Approval
Exemption for research useExemption for research use
-amendment of Patent Act-amendment of Patent Act
Drug labeling excluding from copyrightDrug labeling excluding from copyright
-amendment of -amendment of Pharmaceutical Affairs ActPharmaceutical Affairs Act
Data exclusivity for new indication?Data exclusivity for new indication?
37

38
Future perspectivesFuture perspectives

39
Future perspectives in Enhancing Drug Quality
Strategies for API and Generic Drug
Strategies for GMP regulation
Strategies for Post-Approval Changes

40
Establishing the review strategies for DMF of API and CTD of Generic Drugs Time-line and action plans
to implement API’s DMF and CTD for Generic Drug
Implement DMF and CTD by stages.
Stage 1: With NHI drug price incentives, encouraging companies to
implement DMF, starting from Oct. 1st, 2009
Stage 2: Compulsory implementation of DMF and CTD
-Implement by stages, by items
Strategies for API and Generic DrugStrategies for API and Generic Drug

41
New chemical entities
New biologic
New indication
New dosage forms
New route of administration
Generics
OTC
EU FDA MHLW
included
included
included
included
included
included
included
included
included*
included
included
included
included
included
included
included
included
included
included
not included
not included
* with the exception of blood and blood components
Common Technical Document (CTD)Implementation Coordination Group
presented in June 13 `02General Information on the CTD
organized by:Implementation Coordination Group Members
plus members in CTD-Q, CTD-S, CTD-E & eCTD
Implement Common Technical Document for Implement Common Technical Document for generic druggeneric drug

4242
Regulation strategies for post-approval changes
Establish DMF database of API
Monitor and inspect API changes
Revise guideline for Scale-Up and Post
Approval Changes (SUPAC)
Strengthen regulation on Post-Approval
Changes-Product Quality ReviewRegulation Strategy for management of product
license
Strategies for Strategies for Post-Approval ChangesPost-Approval Changes

43434343
Strategies for GMP regulationStrategies for GMP regulation
Quality Assurance for Drug ManufacturingQuality Assurance for Drug Manufacturing
Good Manufactory Practice (GMP)
Documentations, SOP, QC, QACurrent GMP (cGMP)
Validations- analytical method, process, data treatment
PIC/S GMP by 2014
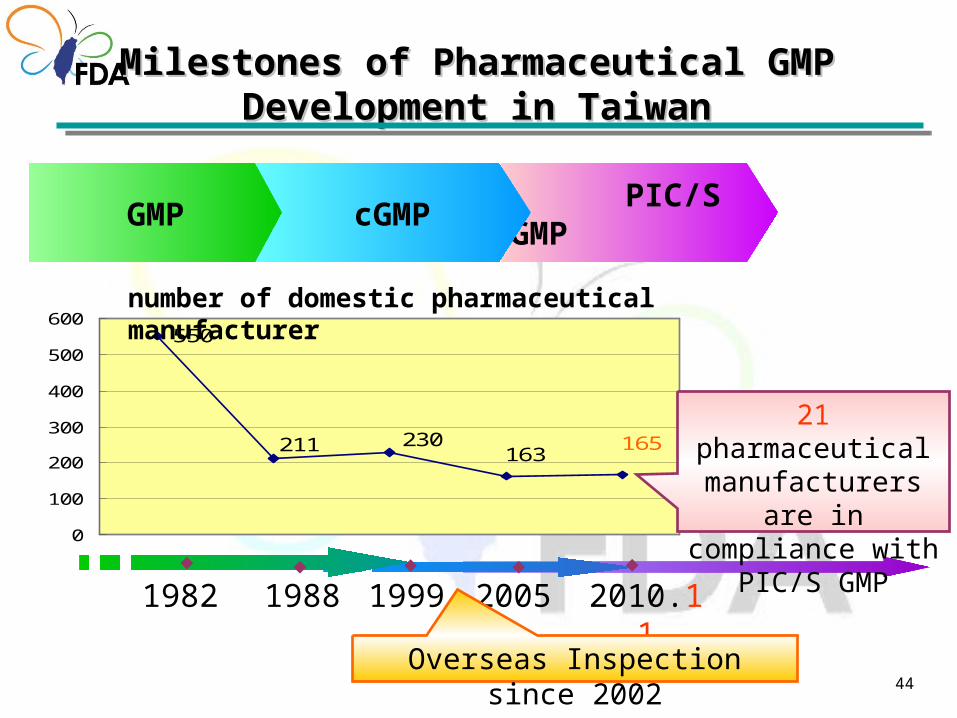
44
PIC/S GMP
1982◆
1999◆
cGMPGMP
◆
2010.11◆
1988◆
2005
211 230163
165
550
0
100
200
300
400
500
600
21 pharmaceutical manufacturers are in
compliance with PIC/S GMP
Milestones of Pharmaceutical GMP Development in Milestones of Pharmaceutical GMP Development in TaiwanTaiwan
number of domestic pharmaceutical manufacturer
Overseas Inspection since 2002

45
Internationalize quality guidelinesInternationalize quality guidelines
Implementation status of ICH quality Guidelines in Taiwan
Q1 Stability Adopt
Q2 Analytical Validation Adopt
Q3 Impurities Accept
Q4 Pharmacopoeias Accept
Q5 Quality of Biotechnological products Accept
Q6 Specifications Accept
Q7 Good Manufacturing Practice(原料藥GMP)
Adopt
Q8 Pharmaceutical Development Accept
Q9 Risk Management system Accept
Q10 Pharmaceutical Quality system Accept

46
Quality by
Testing- Quality Control
Quality by
Manufacturing - Quality
Assurance
1970s 1980s 1990s 2000s
Quality by
Design
Quality Systems
Evolution of Quality conceptEvolution of Quality concept

47
New Quality InitiativeNew Quality Initiative
How to do What to do
Product
-1970s
QualityControl
Process
1980s~1990s
Systems
21st Century
QualitySystems
QualityAssurance
Fixed controls state Dynamic controls state

48
The three-way winThe three-way win
Consumer
Industry Government
Ensure Drug quality,
safety& efficacy
International harmonization
on drugmanagement
Increase international
competitiveness
GoalGoal

49


![VeryLowRateScalableSpeechCodingthroughClassified ...ehsanj/publications/EURASIP_10.pdfEmbedded quantization was first introduced by Tzou [1] for scalar quantization. Tzou proposed](https://img.pdfslide.us/doc/110x75/602a50dcd89ce10f9b6fa2d4/verylowratescalablespeechcodingthroughclassiied-ehsanjpublicationseurasip10pdf.jpg)


























