Embed Size (px)
Citation preview

CHAPTER III
7 APPROACH TOWARD STHE
SYNTHESIS TNAPHTHALENE
MOIETY OF NEOCARZINOSTATIN
\--CHROMOPIJOREJ

Chapter III
3.1 INTRODUCTION
Neocarzinostatin (NCS) 1 was first isolated in 1965 by Ishida and coworkers from the
bacterium Streptomyces carzinostaticus. 2 NCS is made up of a 1:1 non-covalent
complex of an extraordinary reactive nine-membered ring epoxydiyne chromophore
(NCS chrome) 1 (Fig 1) tightly bound to a protein known as apo-NCS (KD = 0.1 nM). 3
1
Fig 1
The chromophore was the first isolated member of the so called enediyne class of
antibiotics and was found to exhibit broad spectrum antitumor activity, which acts as a
DNA-cleaving agent initiated by the radical hydrogen abstraction of a deoxyribose
residue. 4 '5
When not bound to apo-NCS, the chromophore is highly reactive particularly towards
nucleophilic reagents. 6 Nucleophilic activation of the chromophore principally with
thiols,7 initiates a sequence of reactions terminating in the formation of a biradical
species8 that can function as a DNA-damaging agent in vitro.
Scientific interest in NCS stems primarily from its highly unusual chemical composition
and its reactivity, notably the ability to cleave double stranded DNA by a novel
112 'Page

Chapter III
mechanistic pathway. 4 NCS has shown some efficacy in the treatment of human cancers
of the bladder, 9 stomach ic) and liver. 11
The effects of NCS on the growth of mammalian cells in culture are profound. 2 HeLa
cells treated with NCS (z: 20 nM) show delayed entry into and prolonged progression
through S-phase and do not undergo G2-M transition. 12
A polymer-conjugated version of the drug (conjugated via its binding protein), has been
approved for the treatment of cancers of the liver and brain, as well as leukemia, in
Japan. 13
2-Hydroxy-7-methoxy-5-methylnaphthalene-1 -carboxylic acid 2 is a component of
NCS chromophore 1 and is found to play a key role in the binding of 1 to DNA by
functioning as an intercalating group. 14 It has also been shown to play a key role in the
binding of 1 to the neocarzinostatin protein component, lying deep within a cleft in the
chromoprotein complex. 15
Thus, NCS chromophore 1 has been on high demands for the clinical trials as well as
for the total synthesis. The total synthesis of 1 would obviously require easy availability
of its components, for which we undertook the synthesis of one of its important
component; the naphthalene moiety 2.
3.2 LITERATURE REVIEW
The naphthalene moiety of 2 has four different functional groups; therefore, it is a very
challenging task for an organic chemist to prepare it in just few steps from
commercially available starting materials. The methods reported previously are
summarized below.
Shibuya et al. 17 in 1984 revised the previously reported structure 16 of naphthalene
moiety by its synthesis. They started with lithiation of substituted o-toluamide 3 and
obtained the product after seven steps of different functional group interconversions
(Scheme I) in 3% overall yield.
. 11311) age

CONMe 2
OCH 3 0 0 LDA, THF, -78 °C 3.-
0.5h, 98% H3C
0
HO
CONMe 2
OCH3
H3C CHO
H3C
3
CONMe 2
OCH 3 1. Cr0 3, Py, Ac 20
CH2 Cl 2 , RT, 2h, 83%
2. aq. HCI, acetone, RT H 3C 3h, 88%
1. conc. H 2SO4, RT, 12h, 45%
2. K 200 3, S0 2(CH 30) 2
Cat (C4 H 9)4NHSO 4, heat
1h, 92%
CN H 3CO 1. KOH, aq EtOH
2. AcCI, Me0H 93%
H 3CO
CH 3
COOMe
1. LDA, CH 3CHO
2. (COCl 2) 2 , DMSO, NEt 3
86% CH3
Chapter III
OH
CONMe 2 H3CO
OCH3 1. FSO 3CH3, RT, 1h, 100% H 3 CO
50°C, 10min, 31% CH3
CH 3
COOH
1. 10% NaOH, H 2O-DMSO (1:1), 120 °C, 1h, 99%
2. BCI 3 , CH 2Cl 2 , -78°C
0.5h, 87%
H3CO OH
CH3
Scheme I
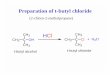
In 1989, Shishido et al.' 8 reported a synthesis of methyl ester of neocarzinostatin moiety
from 1-cyano-5-methoxy-3-methylbenzocyclobutene 4 employing an electrocyclic
reaction of E-o-quinodimethane as a key step (Scheme II).
4
1N NaOH, CH 3CN,
114 Page
5 6
H3C 180°C
o-dichlorobenzene
CH3 CH3
COOMe
E
OEt H3C0
K2CO3
aq. Me0H 97%
CH3
COOMe
H3CO DDQ
quantitative yield
COOMe COOMe H C OEt
H3C0 0 CH3 LDA, cia:iEt
80%
Chapter 111
Scheme II
Overall yield of this sequence was 51% from 4. Another sequence of reaction was
carried out in which 5 was treated with t-butyldimethylsilyl trifluoromethanesulfonate
to provide the silyl enol ether derivative of 6 instead of enol carbonate derivative. The
product was prepared using same reaction conditions in 46% overall yield from 5.
Citterio et a/. 19 reported an application of intramolecular homolytic alkylation of
aromatic compounds by oxidation of methylene group of 1,3-dicarbonyl compounds by
high-valent metal salts. Thus, a series of 2-hydroxy-l-naphthoic acid derivatives were
synthesized by four-electron oxidation of 5-aryl-3-oxopentanoic acid derivatives 8 by
manganese(III) acetate in acetic acid or CAN in methanol, through cyclic intermediates
9, and 10 (Scheme III).
For 2a - an ester of the naphthoic acid, yield using CAN was reported as 62%, whereas
manganese(III) acetate gave several byproducts along with 2.
115 Page

COY
8 9
COY
CAN, Me0H, 20°C
OR Mn(OAc) 3 , AcOH, 40°C
2Mn+
HA
11
silica gel
benzene, heat
H 3CO 1-methylnaphthalene
53%
H 3CO
5% Pd/C, reflux, 30min
13
n-Bu 4NBr3, CH2Cl 2 :MeOH (1:2)
0°C, 2h, 79% OH
12 '
Chapter III
Scheme III
Takahashi et al.2° published a six-step synthesis of 2-hydroxy-7-methoxy-5-
methylnaphthalene- 1-carboxylic acid from 5-methylcyclohexane-1,3-dione 12, using
Robinson annulation reaction as a key step in 23% overall yield (Scheme IV).
HC(OMe)3 , TsOH
Me0H, RT, 20h, 95% H3CO
LDA, -78°C, THF, 40min
3-Buten-2-one, 80%
116 'Page

CH3
H3CO
1. t-Butylacrylate, Et3N, P(o-tol)3
Pd(OAc)2, 108 °C, 89%
2. TFA, CH2C1 2 , 23°C
quantitative H3CO
0 CH3 53 - 63%
14
1. carbonyldiimidazole, THF, 23°C
2. Magnesium methyl malonate
THF, 23 °C, 78% H3CO
CH3
S 02CI 2, C6 H6, 60°C
OH hv, Et3N, CH3 OH
92% CI
H3 CO
H3CO
NaOH, MeOH:H20 (3:1)
H3 CO OH
Chapter III
H 3CO
COOH
n-BuLi, THF, -50 C
CO2 , -30°C, 1h, 88%
° H3CO
O. OH OH
CH3
CH3
Scheme IV
Myers et a/. 21 reported a six-step synthesis of naphthoic acid component employing 4-
bromo-3-methylanisole 14 as a starting material that proceeds in 31-37% yield. The key
feature of this synthesis is photocyclization reaction (Scheme V).
Scheme V
Gorth et a/. 22 synthesized the naphthalene moiety on the ten gram scale in nine steps
and in 43% overall yield from 3,5-dimethylanisole 15 (Scheme VI).
117 'Page

H 3CO CH 3
NaCN, DMF, 90°C
2h, 29% H 3CO
Br P(o-toly1) 3, Et 3N, toluene, sealed tube,
165°C, 16h, 91%, 94:6 (E2) CH3
Ethyl acrylate, [Pd(PPh 3 ) 2]C1 2 ,
H 3CO H 3 CO
C H3 CH 3
1. BrCCI 3 , DBU
RT, 12h, 79% 3.- 2. KOH, MeOH:H 20 (3:1)
70°C, 12h, 89%
Chapter III
NBS, CH 3CN, 0°C to RT NBS, AIBN, CCI 4
H3CO CH3 12h, 100% 70°C - reflux, 2h, 87%
15 H3CO
H 3CO
COOEt Mg turnings, Me0H
sealed tube, 0°C RT, 3h, 96%
H 3CO
COOMe
Conc. H2SO4 , Me0H
COOMe LiHMDS, THF, -78 °C
140°C, 12h, 98% H 3CO
30min. 89%
Scheme VI
Recently Ji et al. 23 described a four-step synthesis of naphthoic acid 2 in which the key
transformation was to prepare Z-selective olefin by coupling reaction between aromatic
aldehyde 16 and phenylphosphonate ester 17 using DBU as a base (Scheme VII).
118 'Page

16
CH3
17
DBU, THF, 0 to 23°C, 82%
H 3C0 H 3CO
CHO
0
CH3 CH3
1. CF3CH 2 OH, NaH, THF, 23C H3CO
2. Mn(OAc)3, HOAc, 23°C, 93% 93% (two steps)
OH H3CO
LiOH, THF, H 2O
40°C, 100% lw
Chapter III
Scheme VII
1191Page
BnO OEt
BnO BnO BnO OH
19 CH3 CH3
20
BnO
CH2
OEt
Chapter III
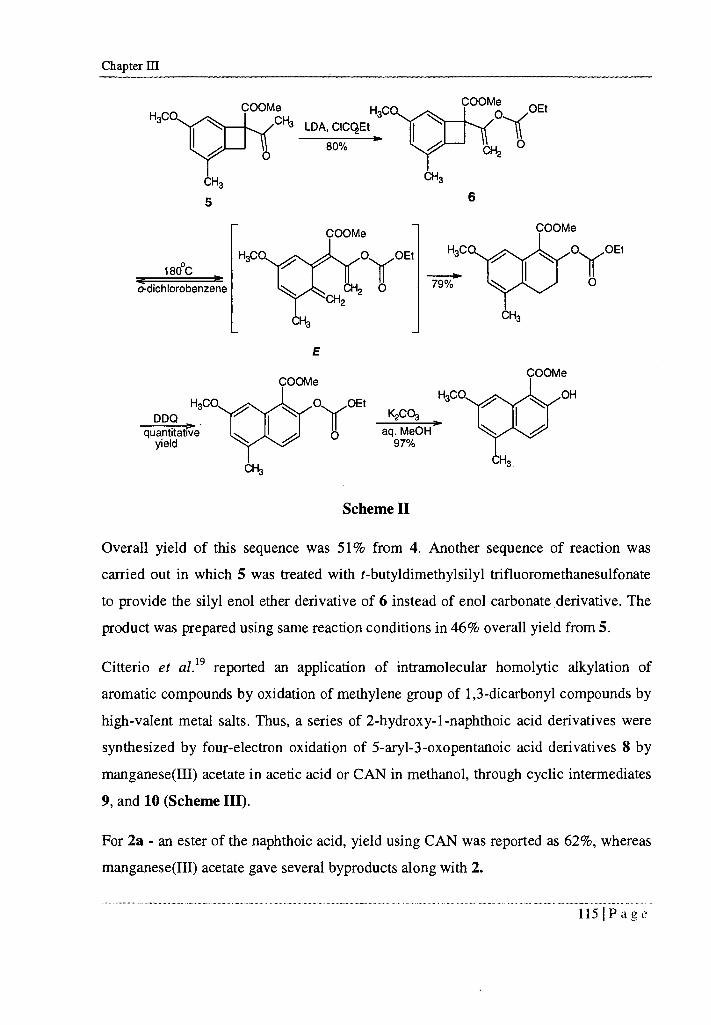
3.3 RESULTS AND DISCUSSIONS
2-Hydroxy-7-methoxy-5-methylnaphthalene-1 -carboxylic acid 2 is rather a complex
molecule due to different functionalities at different positions on the naphthalene ring.
This makes it an interesting target of synthesis.
In our laboratory, several attempts were made to synthesize 2 using Wasserman
chemistry (Scheme VIII).
COCI Ph3 P=CHCO 2Et
H 3 CO
H 3 CO
COOEt H +
H3 CO
H3CO
18
Scheme VIII
In this case, 18 was successfully prepared but several attempts towards its cyclization
failed hence the approach was discontinued.
The next strategy that was used, involved synthesis of naphthalene skeleton 19 first and
then the functional group transformation of the ester to alcohol and further to methyl
ether (Scheme IX).
120 Page

CHO BnO
CH3 22
CH3 CH3 21
COOH
HO HO OCH 3 OCH 3
CH3 CH3
2
OH BnO BnO CH3
BnO CH3
C H3 CH3
22 23 24
BnO CHO (Et0) 20P
25
Chapter III
COOH
OCH 3
Scheme IX
This method was discontinued, as the naphthaldehyde 21 could not be converted to
naphthol 22 by any of the attempted methods.
Here we have made attempts to synthesize naphthol 22 via acetonaphthone 23 which we
thought would easily undergo Baeyer-Villiger oxidation. Accordingly, our
retrosynthetic analysis was as shown below (Scheme X).
Scheme X
We thought that, if 22 is prepared then we can synthesize 2 using the literature method 20
(Scheme IV). 22, in turn Can be obtained by Baeyer-Villiger oxidation of 23.
Isomerization and cyclization of 24 would give 23, whereas 24 can be assembled from
the aldehyde 25 and phosphonate 26 via Horner-Wadsworth-Emmons reaction (HWE).
121 IPage

Chapter III
We started with the preparation of HWE reagent 26. When Arbuzov reaction24 was
attempted between triethyiphosphite and chloroacetone at 80 °C for 3.0 h, we found two
distinct spots on TLC as expected (Scheme XI).
CH2
P(OEt)3 CH3 H3 C0
CH3 PO(OEt)2 PO(OEt)2
27 28
Scheme XI
The separation of the two using column chromatography gave a major product that
showed a strong band at 1661.17 cm -1 in its IR spectrum (neat) and the minor product,
approximately 5% was showing band at 1715.43 cm 1. This proved that the major
product was 28 and not 27, which was not required. We therefore thought of using the
same method that was used for the synthesis of phosphonate of hexanoyl chloride in
previous chapter 25 (Scheme XII).
0
(Et0) 20P
OEt MgC1 2 , TEA, Toluene
CH3COCI (30) (Et0)20P
CH3 Ts0H
0
CH3
P0(0Et) 2
27
O 29
H2O
OEt
0
31
Scheme XII
Thus, when acetyl chloride 30 was added to the cooled solution of 29 in toluene with
MgC12 and TEA, the thick orange colored precipitate was formed, which was further
treated with dilute acid and the product 31 was extracted in diethyl ether.
31 in its IR spectrum (neat) showed bands at 1708 cm -1 and 1737 cm -1 (shoulder).
Without further analysis, 31 was subjected to decarboxylative hydrolysis using catalytic
amount of p-toluenesulphonic acid (PTSA) in water to give crude 27 after 24 h of
reflux. 27 when extracted from water using ether and then with ethyl acetate gave only
122 IPage

Chapter III
63% of the crude product. The crude product when loaded on the column gave only
57% of the pure product from 31 and just 53% from 29 in comparison to that of 72.15%
of diethyl-2-oxoheptylphosphonate in chapter I.
We therefore modified the procedure. Here we took triethylphosphonoacetate 29,
MgC12 and toluene in the round bottom flask and added slowly TEA over a period of 15
min. Stirred for considerable amount of time at room temperature for the formation of
an anion. This anion was then trapped by slow addition of acetyl chloride. After the
confirmation of absence of 29 by TLC, dilute HC1 was added slowly to the reaction
mass and continued stirring for 1.0 h more. Extraction of the product in toluene and
refluxing the toluene layer for 16.0 h in presence of catalytic amount of PTSA gave us
almost pure crude product, which was filtered through the column to obtain 77.8% of
pure 27 as a colorless liquid. Boiling point of 27 was 93 °C (lmmHg) [lit26 by =
98°C/lmmHg]
This product 27 in its IR spectrum (neat) showed band at 1715.76 cm"' implying the
presence of keto-carbonyl.
In its 'H NMR (CDC13, 300 MHz, 6 ppm), (Fig 2) spectrum 27 showed a triplet at 6
1.35 ppm (J = 7.0 Hz) integrating for six protons indicating the presence of two —CH2-
CH3 group. The corresponding multiplet for four methylene protons was seen at 6 4.16
indicating the presence of —OCH2CH3 group. A singlet at 6 2.33 for three protons
indicated the presence of —COCH3 group. A doublet at 6 3.10 for two protons with J =
22.8 Hz confirmed the presence of methylene group between phosphorane and carbonyl
group.
Thus, based on mode of formation and spectral analysis, 27 was confirmed to be
diethyl-2-oxopropylphosphonate.
Diethyl-2-oxopropylphosphonate 27 was then reacted with allyl bromide to obtain its
allylated derivative 26 as viscous oil (Scheme XIII).
123 IPa ge

0
1. NaH, THE
H3C
26
(EtO) 2 0P CH3
P0(0E1) 2
27
2. H 2C=CH-CH2Br
Chapter III
1/1 U) CO V. O Cg r•-1 •cl• VI Cg O eel eel CO 01 CU CO In Cg CO L0 0 CO CO Ln Cg ••••1 4■1 r. CO
eel kD O ,D (el 01 Ca CT 01 CO
V. lt)
eel l0 Cs. C.. lft eel eg ♦ ..1 0 0
L.r> GI• CV V) Pi eel eel
CT SD r1 CV
• • • • V. V. GI• V.
• • V. V.
• r•1 rel rn CS! Cg N CV
• • • N CSO N CV CV ,1 •-1
WY/ I 1111/ N
5.0 4.5 4.0 3.5 3.0 2:5 2.0 1.5 1.0 0.5
O O
Fig 2
Scheme XIII
In its IR spectrum (neat) 26 showed a band at 1715.18 cm -1 probably due to the
carbonyl group.
124 Page

1251 age
5 . 0 4 . 5 4 . 0 3 . 5 3 . 0 2 . 5 2.0 1 . 5 1 . 0
21 rs•
1 11 101
6 . 0 5 . 5
hE:r )01> Fig 3a
Chapter III
1 H NMR (CDC13, 300 MHz, 8 ppm), spectrum (Fig 3a) of 26:
6 1.35 t (J = 6.9 Hz) 6H 2 X CH3CH20
6 2.32 s 3H -COCH3
6 2.56 m 1H HCHCH=CH2
6 2.76 m 1H HCHCH=CH2
6 3.26 m 1H PCH
6 4.15 m 4H 2 X CH3CH20
6 5.02 — 5.17 m 2H CH=CH2
6 5.73 m 1H CH=CH2
CO 0 IV •cl• .•-I 0 I... I... 0 CcI PI P 01 Is.- cr Cc/ C,/ co m 0 an rc m an co an in Ott cr cv m M in cr kJ:, co rc ma M r 0 co M 0. CcI ...I 01 CO Cs- In II -- .-I0 in CV 0 oo, , ,... r■ ,e, , c., 0 0 N. 0 c.. IV en 0 01 Cc, OS 0 CV 0 , 0 .4 • CV 0 Iss 0 •0 . r-o , c-I 0 C.1 v-I ..-I 1-1 .1 I r-1 in on M C. N C. C. N.- co r r r r M M in in 0
an ta, in an an an m an an an an an an an an cp. .3. .3. cr cr cr .3. .ar , rn on in cm ro ro m m co m ol NN cv C.1 CV CV NN
,0 CV 01 CcIMMC•1,1(N CO
I-1 c-1
\ ////

100- 357.0917
1Calcd Mass = 257.0919 (M+Naj
179.0454 207.0704 289.1115
1
i ( 1
. , i 0 . . . , .-, 1.. , ■ . . ..11--t. 1)1,44 6 4•:7 , i••ty'.. , 4 :1 1, , 4, ...3. f "..I' ,. 1—,-0,- -.. 4, ,---,-4,-.4-- . I. . 1 , • . , i• -- , ,• -, •,--, , , m/z
inn 125 isn 175 Inn 225 250 275 1nn .-42 15n 375 400 425 450 475 500 525 550 575 600 625 650 675 700
Chapter III
The high resolution mass spectrum (HRMS) of 26 (Fig 3b) having strong peak m/z
257.0917 was presumably due to the (M + Na) + pseudo ions.
The elemental composition of which was determined to be C101-11904P. HRMS; m/z
calculated for C101-11904PNa [(M + Na)}+ = 257.0919, found = 257.0917.
Fig 3b
The yield of 26 was found to be 74.52%.
Thus, on the basis of mode of formation and spectral analysis structure, 26 was assigned
to be diethyl (2-oxohex-5-ene-3-yl)phosphonate.
The phosphonate 26 was then subjected to HWE reaction conditions with 3-
benzyloxybenzaldehyde 25 to obtain 24 (Scheme XIV).
The IR spectrum (neat) of 24 showed a band at 1667.53 cm', probably due to the
carbonyl stretching. The lower value must be due to the conjugation.
126IPage

Chapter III
BnO CHO
(Et0) 20P
BnO H3
25
H3C
26
24
Scheme XIV
NMR (CDC13, 300 MHz, 6 ppm), spectrum (Fig 4a) of 24:-
6 2.48 s 3H -COCH3
6 3.28 br s 2H -CH2CH=CH2
6 5.01 br s 1H -CH2CH=CHH
6 5.07 br s 1H -CH2CH=CHH
6 5.10 s 2H PhCH20-
o 5.97 m 1H CH=CH2
6 7.62 s 1H CH=CCO
6 6.83 — 7.44 m 9H ArH
The high resolution mass spectrum (HRMS) (Fig 4b) of the compound having peak at
miz 315.1361 was presumably due to the (M + Na) + pseudo ions.
The elemental composition of 24 was determined to be C20112002. HRMS; nilz
calculated for C20H20O2Na [(M + Na)+] was 315.1361, found = 315.1361.
Thus, on the basis of mode of formation and spectral analysis, structure 24 was assigned
to it.
Yield of the product 24 obtained was found to be 65.7%
127 P a g

7.5 7.0 6.5
6.0
5.5 5.0
s— ea pi I
Fig 4a
3.5 3.0 2.5
tr, C41
4.5 4.0
Chapter 11.1
OCCO•11-•1...INC00,01f0.4 . ..O-O.NO
OCO11.103,...flO)Oc0 (0 N 1 - 01 el ,-4 01, Lt.,' el
tosrem•linel•I•IN000 ■TO, 0 0,3,1). CA . . . . . . • • • . • • • • . . • •.,..:
O.O.O..O.,O.O-O-O-C,O.O.... 10O1O101.11.
1001 lealcd Mass = 315.1361 (M +Na 315.1361
)
257.0905
413.2672
16.1388
, ),, , • rl... ,,,, 100 150 200 250 300 350 400 450 500 55U
• • • r MiZ 600 650 700. 750 800 850
Fig 4b
128Ipage

Chapter III
After successfully obtaining the HWE product 24 in sufficient amount, it was then to be
annulated to 23. Initially, we tried Pd/C, which was used earlier in our group for the
synthesis of carbazole 27 (Scheme XV).
BnO CH3
Pd/C
BnO CH 3
nitrobenzene reflux
CH2 24
CH3
BnO CH3
BnO CH3
CH3
23
CH3
Scheme XV
It was expected that first isomerization of the allylic double bond would take place,
followed by electrocyclization to get dihydronaphthalene which would then, under these
reaction conditions, aromatize to the required naphthalene 23. However, though TLC
indicated consumption of the starting compound, there were too many spots on TLC to
effect separation of any product.
Next, we attempted the use of base for the isomerization of the double bond. The base
used was KOH in refluxing DMSO. However, no change was observed in the starting
(TLC). Lastly, we tried well known RhC13.X H 2O for isomerization in refluxing
ethanol. In this case, it lead to a complex mixture.
Having failed to achieve annulation by isomerization of the double bond, we thought of
electrophilic substitution reaction as shown below (Scheme XVI).
129 Page

CH3
CH3 Bn0
CH3
CH3
Bn0 CH3
CH3
[0]
CH3
23
Chapter III
Scheme XVI
It was speculated that after electrophilic substitution the dihydronaphthalene formed
would get air-oxidized to naphthalene 23. We tried two acids viz. triflouroacetic acid
and sulfuric acid for cyclization.
Thus, 24 was refluxed in TFA till disappearance of the starting materials on TLC.
Purification of the reaction product by column chromatography gave a light yellow
solid. The melting point of the solid was found to be 192 — 194 °C.
In its IR spectrum (KBr) the product showed strong bands at 3178.83 and 1653.07 cm -1 .
1 H NMR (CDC13, 300 MHz, 6 ppm), spectrum (Fig 5a) of this compound showed:-
6 2.45 s 3H ArCH3
6 2.73 s 3H -COCH3
6 4.54 s 2H -CH2Ph
0 5.14 . s 1H -
(5 7 .19 — 7 .30 m 6H ArH
0 7.74 s 1H ArH
130 Wage

1400
7
H 6 dd (meta,ortho coupled)
singlet m (5H)
\ PhH2CO
H
8 singlet CH3
CH 3
Chapter III
6 7.92 d (J = 9.3 Hz) 1H ArH
6 8.48 s 1H ArH
13C NMR (CDC13, 75 MHz, 6 ppm) (Fig 5b) of this compound showed 6 19.80T
(ArCH3), 27.09T (COCH3), 30.531 (CH2Ph), 120.71T (CH), 120.99 (Cq), 121.22T (CH),
124.33T (CH), 124.48T(CH), 126.06T (CH), 128.56T (CH), 128.71T(CH), 129.28 (Cq),
133.11 (Cq), 134.22 (Cq), 135.58 (Cq), 141.86 (Cq), 153.55 (0-Cq), 198.52 (CO).
The required product 23 was expected to show two singlets for the two methyl groups,
(ArCH3 and COCH3), one singlet at about 6 5.1 for the two protons of the benzyl group
(OCH2Ph), five aromatic protons of benzyl group and five different aromatic protons of
the naphthalene ring as shown below (Fig 6).
d (meta coupled)
d (meta coupled)
d(ortho coupled)1
d (meta coupled)
singlet
Fig 6
As expected, the product using TFA showed two singlets at 6 2.45 and 2.73 ppm that
we could assign for the two methyl groups Ar-CH 3 and COCH3 respectively. A singlet
appearing at 6 4.54 ppm for the two protons was of methylene of benzylic group but it
was appearing slightly upfield than expected. The singlet at 6 8.47 ppm could be due to
C1-H proton of naphthalene ring getting deshielded due to adjacent carbonyl group. The
131IP a g

Chapter III
second singlet at ö 7.73 ppm could be due to the C3-H of the naphthalene ring and the
doublet could be due to the C5-H of the naphthalene ring. The C6-H and C8-H protons
must be merged with the aromatic protons of the benzylic group. However, integration
indicated that there is one less proton. Also the singlet at ö 5.14 remained unaccounted.
Considering this data, we assayed the following structure to the product 23a (Fig 7).
HO CH3
CH3
23a (Fig 7)
The singlet at ö 5.14 could be due to the hydroxy group which was also indicated in IR
spectrum by the band at 3178.83 cm -1 . The band may be at higher frequency due to
intermolecular hydrogen bonding. Further the upfield shift of the benzylic methylene
protons (6 4.54) could now be accounted, as it was not attached to electronegative
oxygen. This was confirmed by appearance of its carbon in 13C NMR at ö 30.53 ppm.
The high resolution mass spectrum (HRMS) (Fig 5c) of the compound displayed a
strong peak at m/z 313.1218 that was presumably due to (M + Na) + pseudo ions. The
elemental composition of which was determined to be C20111802, HRMS; m/z calculated
for C2:J-11802Na [(M + Na) +] was 313.1204, found = 313.1218.
The yield of the compound was 58.4%.
Simultaneously, we also tried a ring annulations reaction using 40% of aq H2SO 4 in
DCM. This reaction was worked up after three days (TLC still indicated presence of
starting). The product 23, which was obtained here displayed higher Rf than the starting,
which was different from TFA product (product Rf was lower than starting). The light
yellow solid melted at 102 — 106 °C.
The IR spectrum (KBr) of 23 showed a carbonyl band at 1680.00 cm -1
132IPage

intramolecular alkylation
CH3
23a
CH3
CH3 23
Chapter III
NMR (CDC13, 300 MHz, 6 ppm), spectrum (Fig 8a) showed a singlet for six protons
at 6 2.72, which can be, attributed to the two methyl groups. A singlet at 6 5.24
integrating for the two protons can be attributed to the benzylic methylene. Multiplets
from 6 7.36 to 7.53 integrating for the seven protons can be attributed to the aromatic
protons. Two singlets at 6 7.75 and 8.23 for single proton each can be attributed to the
two protons at C, and C3 position of naphthalene ring. The doublet at 6 7.97 (J = 9.0
Hz) can be assigned to the protons at C5 of naphthalene ring.
The high resolution mass spectrum (HRMS) (Fig 8b) of the compound displayed a
strong peak at m/z 313.1223 was presumably due to the (M + Na) + pseudo ions. The
elemental composition of which was determined to be C20111802. HRMS; m/z calculated
for C201-11802Na [(M + Na) ±] was 313.1204, found = 313.1223.
Thus on the basis of mode of formation and spectral analysis we assigned structure 23
for it. This was further confirmed by the following 13C NMR.
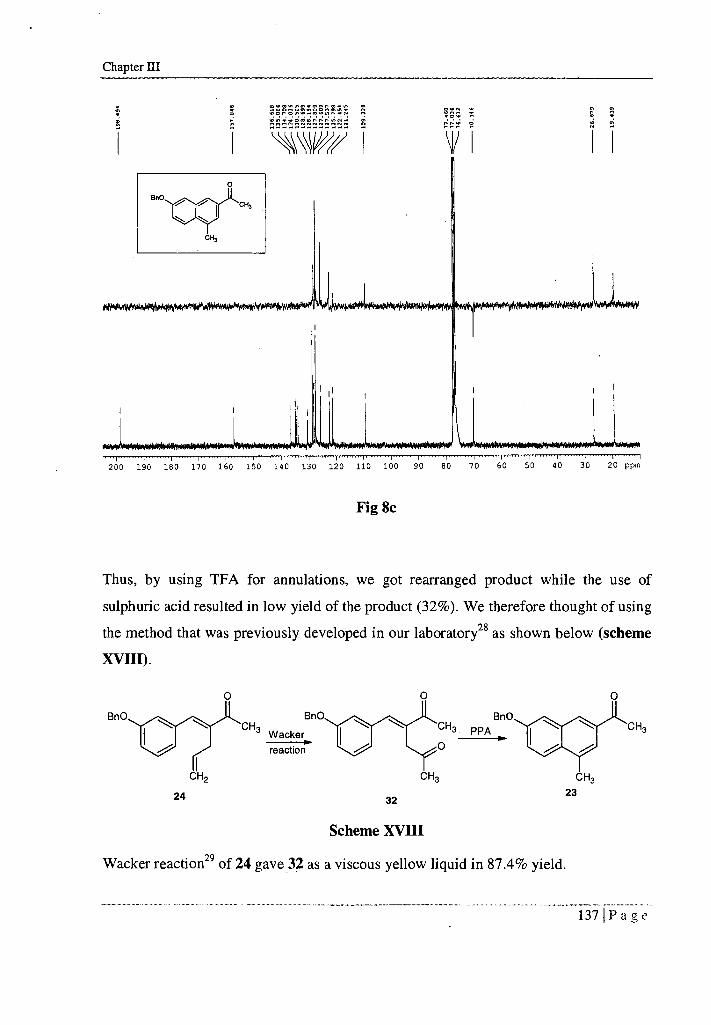
13C NMR (CDC13 , 75 MHz, 6 ppm) spectrum (Fig 8c) showed 6 19.441 (ArCH3 ),
26.601 (CH3CO), 70.151 (OCH2Ph), 109.341 - 128.681 (ArH), 134.02 (Cq), 134.68
(Cq), 134.98 (Cq), 136.60 (Cq), 157.02 (Cq - COBn), 198.47 (CO).
The multiplicities of the carbon signals were obtained from DEPT 135 experiment.
It appears that the product 23 formed under the reaction conditions got rearranged to
23a by intramolecular alkylation as shown below (Scheme XVII).
Ph .
Scheme XVII
133 IPage

ut co r•-■ NN rn
Chapter III
To confirm formation of 23a, via above postulated mechanism compound 23 was
refluxed in TFA. Gratifyingly, we obtained 23a as expected.
, .0 , CO.CONCOO , 1,1 P 0 , cl• ...I I..' 1 01 CO ,C, ul r 4 01 ,..., o• .-101 •o• Ch 01 r• NNNNN. . to NV
• • • 0 r r- r r r r r r r tr) v rn el
V N/
8.5 8.0
O O
)„
(N) 0
7.5
r-1
7.0 6.5 6.0 5.5 5.0 4.5
D O
4 . 0 3.5 3.0
01 0
A) O s.0
C•ri
2.5
Fig 5a
134 a g.e

.1 ppm 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
1001 Icalcd Mass = 313.1204 (M±Na)!
345.1411
377.:1625
m/z 160 -' 125 - 150 155 200 225250 255 300 325 350 375 400 415 450 475 500 '-'526 56 575 600 625 650
313.1218
Chapter III
Fig 5b
Fig 5c 135 IPage
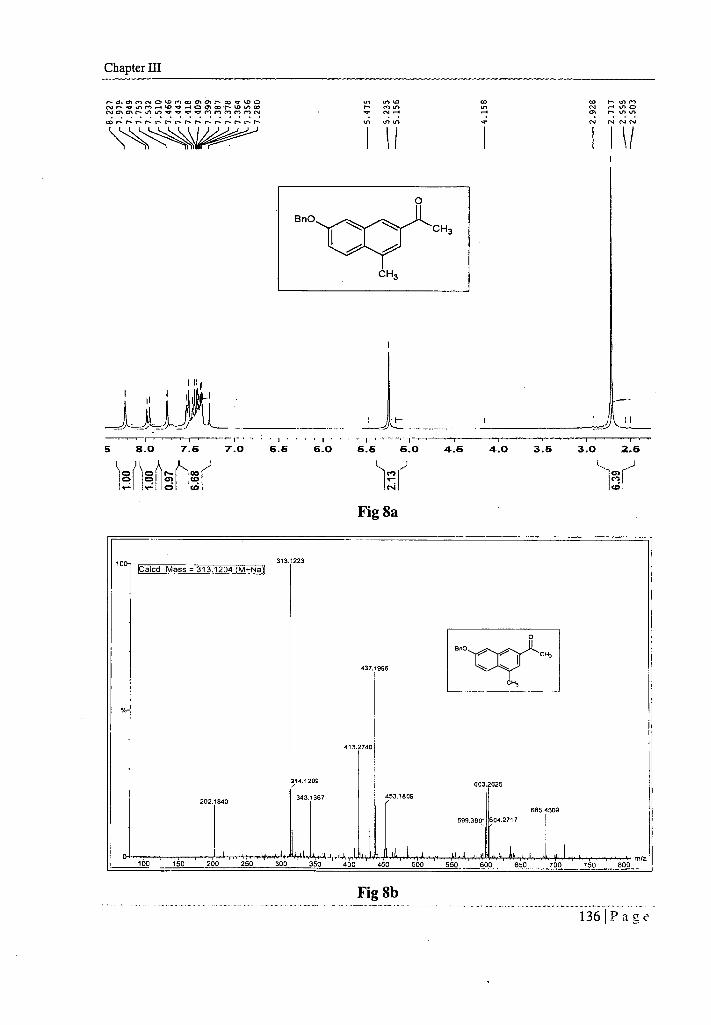
Ln VJ 01 111
•cr CV
U1 1.0
r 01 01 nl ('JO 01 CO ON t•••• v• (D O NJ N VP tr. In cr Ql ar. 1-- 10 to co 0401010- 03 in •cr V•V•1111,11,10IN(V
cg;• • • • • • • • • • • • • • •
r N N N r r N r
4.0 3.5 7.0 6.6 6.0 6.6 6.0 4.6 3.0 2.5
!cal
5 8.0 7.6
c2 (1 c2)N.)
<—
oo 1(0)
cri Ict. 1 co
Chapter HI
1
Fig 8a
100- 313.1223
ICalcd Mass = 313.1204 (M -.-Na)1
437.1996
0
Bn0 CH,
O.
CH,
202.1840
i :1+.-;,4•
!314.1299
343.
,A.
413.2740
367
1114-if 14, T,
453.1809
599.3901
603.2625
685.4509
604.2717 1
, --s, 1. • ; f mtz 100 150 200 250 300 350 400 450 500 550 600 650 ..... 700 750 800
Fig 8b
136 Page
BnO Bn0 CH3
Wacker
reaction
23
CH3 CH3
C H 3 Bn0
CH3 ppA
0
32
Chapter III
200 190 180 170 160 150 190 130 120 110 100 90 80 70 60 50 90 30 20 ppm
Fig 8c
Thus, by using TFA for annulations, we got rearranged product while the use of
sulphuric acid resulted in low yield of the product (32%). We therefore thought of using
the method that was previously developed in our laboratory 28 as shown below (scheme
XVIII).
Scheme XVIII
Wacker reaction 29 of 24 gave 32 as a viscous yellow liquid in 87.4% yield.
137 Pacze

F - cp v. OD el I-1 cc) un 41,1 CV in SO cc, r r rc ni r
•cr cc, CS CO CO el C. 0 0 01 ON ON ON ' Ln
cv cc) CY TV 0
(". C•1
Chapter III
The IR spectrum (neat) of 32 showed two distinct carbonyl stretching bands at 1715.76
cm-1 and 1666.57 cm-1 .
1 11 NMR (CDC13, 300 MHz, 0 ppm), spectrum (Fig 9a) of 32:
6 2.22 s 3H CH3CO
6 2.49 s 3H CH3 COC=
6 3.55 s 2H OCCH2C=
0 5.09 s 2H PhCH2O
66.91 — 7.46 m 9H ArH
67.72 s 1H PhCH=C
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.6
C.,
Fig 9a
4.0 3.5 3.0 2.5 2.0
138 I P a gc

raj-CaMass = 331.1310 (1\4+Na)1 331.1316
32.1420
347.1228
10
Chapter III
The high resolution mass spectrum (HRMS) (Fig 9b) of the compound displayed a
strong peak at m/z 331.1316 was presumably due to the (M + Na) ÷ pseudo ions.
The elemental composition of which was determined to be C20112003. HRMS; m/z
calculated for C20112003Na [(M + Na) ÷] was 331.1310, found = 331.1316.
,• L, h„
_100 150 200 250 300 350 400 450 500 550 600 650 700
Fig 9b
Thus on the basis of mode of formation and spectral analysis structure 32 was assayed
to it.
When cyclization was done using polyphosphoric acid (PPA) at —5 °C, the product
obtained after column purification was same as that of 23, when compared on TLC.
This was further confirmed by IR and 1 H NMR spectra analysis.
Once we had sufficient amount of naphthalene 23 in our hands, we next planned to
carry out Baeyer-Villiger oxidation on this acetonaphthone (Scheme XIX).
1391Pae.e

Chapter III
BnO CH3
Baeyer-Villiger
Oxidation
BnO CH 3
CH3 CH3
23
33
Scheme XIX
We tried the following different conditions:
1. mCPBA in CHC13, reflux - decomposed products
2. mCPBA in CH2C12, RT, N2 atm, 1 week - no reaction
3. a. mCPBA, CHC13; b. NaOH, DMS - 20% starting recovered that was the only major spot
4. mCPBA, grinding — no reaction
5. mCPBA, DCE reflux, Ar atm — decomposed product
6. formic acid, 50% H202, EtOAc reflux - decomposed product
7. formic acid, 30% H202, EtOAc reflux - decomposed product
It appears that, under refluxing conditions either the Baeyer-Villiger product formed is
getting decomposed or acetonaphthone 23 itself is getting decomposed. The failure of
the above strategy prompted us to think of new method towards the synthesis of 2. So
we thought of exploring the azide-isocyanate method that we used for the synthesis of
cherylline dimethyl ether 3° in chapter II. Our retrosynthetic analysis is shown below
(Scheme XX).
COOH
HO OCH3 BnO OH BnO
CH3
CH3
2
22
34
140 Page

Chapter III
BnO
CH2 36
BnO CHO
Ph3P
Scheme XX
This strategy involved Curtius rearrangement 31 of 36 to get isocyanate 35, followed by
hydrolysis to yield an intermediate 34. 22 can then be obtained from 34 by annulations.
We started with the preparation of allyl phosphorane 37 using literature method 32
involving allylation of stable phosphorane 38 (Scheme XXI).
OEt Br
CH2
Chloroform Ph3P
Ph
38
37
Scheme XXI
The allyl bromide was refluxed with stable phosphorane 38 in chloroform for 2.0 h and
the phosphorane salt obtained, was then treated with 2N NaOH to obtain stable allyl
phosphorane 37 as a white solid.
Melting point of 37 was found to be 121 — 122°C (lit. 32 122°C).
The phosphorane was then reacted with 3-benzyloxybenzaldehyde to obtain the Wittig
product 39, which without any further purification was subjected to basic hydrolysis to
obtain the acid 36 as a white solid (Scheme XXII) after acid base treatment.
141 Page

Chapter III
36
Ph3 P
CHO
BnO
37 39
Scheme XXII
36 in its IR sectrum showed a strong band at 1716.65 cm -1 and a broad band at 2750 —
3300 cm-1 indicating the presence of an acid group.
The 1 11 NMR (CDC1 3 , 300 MHz, 6 ppm), spectrum (Fig 10a) of 36 showed a doublet at
3.30 (J = 4.8 Hz) integrating for two protons attributed to the methylene protons next
to double bond. Singlet at 6 5.07 and a multiplet at 6 5.07 — 5.18 integrating for four
protons was assigned to the benzylic methylene group and olefinic methylene group.
Multiplet at 6 6.01 for single proton could be attributed to the methine proton of the
double bond. The two multiplets from 6 6.87 - 7.07 and from 8 7.28 - 7.44 for three and
six protons respectively were assigned to all the aromatic protons. A singlet at 8 7.19
integrating for one proton indicated the olefinic proton on double bond conjugated to
the acid carbonyl.
The 13C NMR (CDC13, 75 MHz, 6 ppm) spectrum (Fig 10b) of 36 showed: 6 31.331
(CH2CH=), 70.041 (PhCH2O), 115.381 (CH2=CH), 115.871 (CH2=CH), 116.031
(CHA ), 122.291 (CHA), 127.401 (CHA ), 128.021 (CHAO, 128.601 (CHA ), 129.471
(CHAO, 129.56 (Cq), 135.271 (CHA ), 136.40 (Cq), 136.71 (Cq), 142.601 (CH=CCO),
158.76 (Cq), 173.19 (CO).
The multiplicities of carbon signals were obtained from DEPT 135 experiment.
The percentage yield of the product was 66.2%; mp was found to be 102-104 °C.
Thus, based on the mode of formation and spectral analysis the structure 36 was
assigned to the acid.
142IPage

Chapter III
e..1 r- e..1 01 e. C' 0 ON N.0 0 NO CV rel CO .-I NO 01 0 CO e-1 01 CO ON
..-1 en .-I CO I-- V, el 0 CO N.0 C' CV 0 e. I-- el .-1 01 I.- V) r- C' .., ON NO
Os C' ne el el el el fi CV 000 ON CO 000 ON 0 C' e, e-I e, 0 0 • - • • • • • • • • • • • • • • • • • • • • • • •
I.- I.- e- e- I-- e.• I.- , I.- r- r- r- to to to to to
CO vl 01 NO e, ON NO fi IN
Cl
I I V
•
Fig 10b
7.5 7.0
ico ) ,7
I GI
6.5 6.0 5.5 5.0
Ifill "el
Fig 10a
3.5 3.1
)4.,V) ccd
4.5 4.0
110 100 90 70 60 50 40 30 ppm 170 160 160 140 130 120
143 IPage

Bn0
36 CH2
Bn0
CH2
Bn0 Bn0
35
Chapter HI
Acid 36 was then subjected to the Curtius reaction conditions to obtain first the azide 40
and then the isocyanate 35, which on hydrolysis would yield 34 (Scheme XXIII).
40
34
Scheme XXIII
Azide 40 and an isocyanate 35 were considered extremely unstable. Therefore all the
steps in above scheme were carried out without analyzing the intermediates.
IR of the crude azide 40 showed a peak at 2139.06 cm -1 and 2262.50 cm-1 , which means
the product had already started rearranging to isocyanate. IR of crude isocyanate 35
showed peak at 2264.43 cm-1 . Both 40 and 35 in their TLC showed many spots. When
acid hydrolysis of crude 35 was carried out, the reaction mass showed multiple spots on
the TLC. When hydrolysis was tried using aq NaOH again there were many spots on the
TLC. We tried a Curtius reaction using classical method as it had worked well in the
case of cherylline. But since isocyanate and azide itself were giving lot of impurities we
thought of using modified Curtius rearrangement method. The results here repeated as
in the previous case so we discontinued this method.
The intermediates formed in both the above methods were highly reactive towards the
reagent present in the reaction mass due to which we ended up with a complex mixture.
We therefore thought of a strategy that would directly lead us to methyl ether of 22 and
1441page

OCH3
CH2
43
HO OCH3 BnO
2 CH3 41 CH3
BnO OCH3 BnO CHO
CH2 42
Ph 3P—\
0—CH 3 2. H2C...=\
Br
Br
Ph 3 P
CI-
+ 1. IBuOK+
O—CH3
tH2
tBuOK+ [Ph3P
1
43
Chapter III
not the hydroxy compound. Thus, our next retrosynthetic approach is as shown below
(Scheme XXIV).
COOH
Scheme XXIV
We planned a synthesis using 1-methoxy-l-triphenylphosphoranylidene-3-butene 43;
which on Wittig reaction with 3-benzyloxybenzaldehyde can lead us to 42. 41 in turn
would be a product of catalytic cyclization of 42, which can then be converted to 2
using the literature method. 2°
The required Wittig reagent 43 was prepared by allylation of the phosphorane, that is
obtained by basification of (methoxymethyl)triphenylphosphonium chloride. The
condensation of 43 with 3-benzyloxybenzaldehyde in situ would lead us to 42 (Scheme
XXV).
/CH2
CH 3
145 Wage

Chapter III
BnO CHO
BnO OCH 3
CH2 42
Scheme XXV
The reaction was tried under different conditions those are mentioned below
Potassium-t-butoxide, THF, N2 atm
n-BuLi, THF, N2 atm
Potassium-t-butoxide, Diethyl ether, N2 atm
Potassium-t-butoxide, THF, Ar atm
Potassium-t-butoxide, THF, Toluene, N2 atm
oxide and triphenylphosphine
oxide and triphenylphosphine
oxide and triphenylphosphine
oxide and triphenylphosphine
Product 42 as oil, 20.64% yield.
The 1 H NMR (CDC13, 300 MHz, 6 ppm), spectrum (Fig 11a) of 42 showed a doublet at
6 4.06 (J = 5.7 Hz) integrating for two protons indicating the presence of CH2CH group.
The two singlets at 6 4.54 and 6 5.11 can be attributed for the four methylene protons.
The two multiplets at 6 5.23 — 5.37 integrating for two protons and at 5.93 — 6.05
integrating for one proton indicated the presence of CH 2=CH group. The nine aromatic
protons were seen as two multiplets from 6 6.93 — 7.05 and 7.27 — 7.46.
The presence of three methylene groups and the absence of methoxy group in the 1 H
NMR indicated formation of 34, and not 42 (Fig 12).
Bn0
Fig 12
1461P a e

Chapter III
This product formed was in very less quantity. We wanted to increase the yield to do
further reactions. When the reaction was repeated several times, somehow, only oxide
and Ph3P was formed. Unfortunately, we were not able to reproduce the process.
,r) r. in .--1 1/40 a) sr/ DI SO 0 0 CO r-C 0 0 SO r-- o cv o r.1 kfl SD V. 01 N Cs) V. 01 OD SON Cfl .0• CV 0 •-1 SO •••T Cr• CV OS N DI (",1 01 CO N .1, 01 0 (11 tfl el (NI 0 CO SO •c/. CSI N . lil •-1 •--1 0 Csi 0 SD N nr 0 CO N tf) 444 tr 4 e• 4-) in D, rr) es: CV DI 0 01 01 01
• • • • • • . • • I 0 0 0 0 001 01 CI DI r'') r") r") (sh CV r-I CO r-. L0 ( CS1 0 0
N r•-• N r- N N N N N r• r- r- SD 1.0 SO • • • • • • • • • • • • • • • • •
k0 SO SD SD ef• al Lrs 0 in tr5 to LC) 1.11 al er, 444 .44 cr • •
ar a' • •
ar .a..
AW I V V
7.5 7.0
.11)1 N
k•-;
6.5 . 6.0 5.5 5.0 4.5 4.0 3.5 , ) ;
•ica i si cr, 01411 pi r: .
C', i I NI
f-r- i I CT2 I !;s 1 N' le4 .-
Fig lla
At this point we stopped all our attempts for the synthesis of naphthalene moiety of
neocarzinostatin chromophore.
147 Page

Chapter III
3.4 CONCLUSION
We have attempted the synthesis of naphthalene moiety 2 of neocarzinostatin
chromophore 1 via substituted acetonaphthone using Baeyer-Villiger oxidation. Our
attempts to synthesize 2 using the Curtius rearrangement method and by using Wittig
reaction of the corresponding aldehyde with unstable 1-methoxy-l-
triphenylphosphoranylidene-3-butene failed to give us the expected results.
148 IP a ge

C H3
CH3
PO(OEt) 2
27
CH2
H3C/L
0
PO(OEt) 2
28
P(OEt)3 heat
MgCl2 , TEA, Toluene OEt (Et0) 20P
CH3COCI (30)
CH3 Ts0H
CH3 H2 O
(EtO) 2 0P
29 OEt PO(OEt) 2
27 O 31
Chapter III
3.5 EXPERIMENTAL
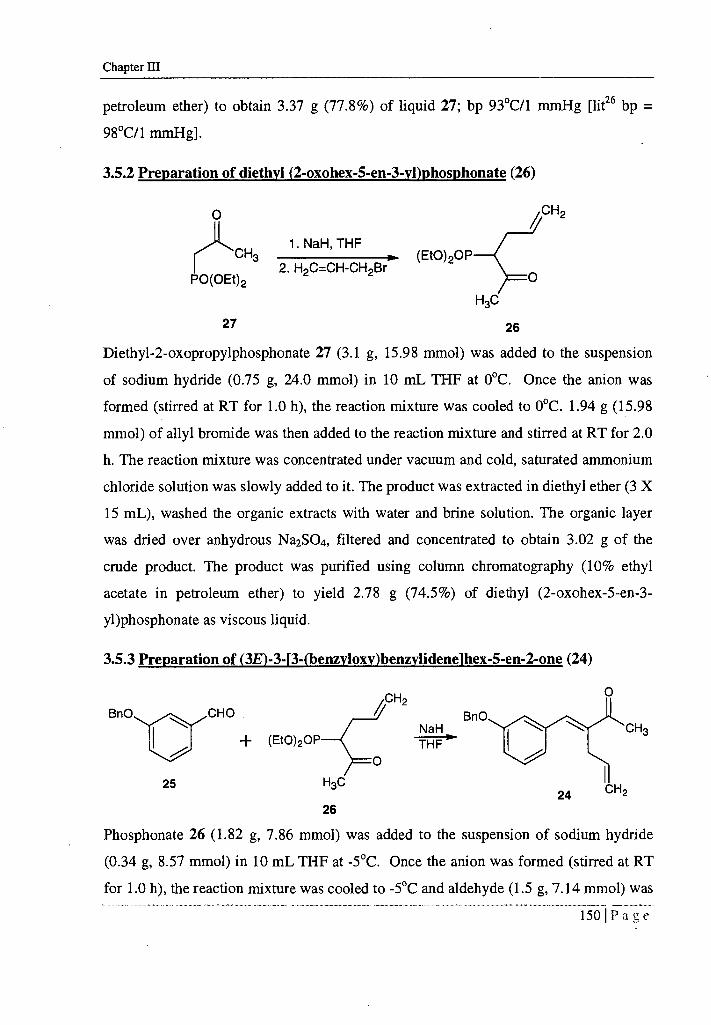
3.5.1a Preparation of diethyl-2-oxopropvlphosphonate via Arbuzov reaction (27)
Mixture of chloroacetone (1.0 g, 10.81 mmol) and triethylphosphite (1.79 g, 10.81
mmol) was heated and refluxed for 3.0 h. Remaining chloroacetone was removed under
vacuum. 1.98 g of the crude product obtained was purified using column
chromatography (20% ethyl acetate in petroleum ether) to obtain 1.64 g (63.3%) of 28
and 0.12 g (4.63%) of 27.
3.5.1b Preparation of diethyl-2-oxopropylphosphonate via acylation of
phosphonate (27)
Triethylamine (5.64 g, 55.84 mmol) in dry toluene (10 mL) was slowly added over a
period of 15 min to a stirred solution of triethyl phosphonoacetate (5 g, 22.32 mmol),
MgC12 (2.13 g, 22.32 mmol) in toluene (20 mL). The mixture was stirred at RT (25 °C)
for 30 min. Acetyl chloride (2.34 g, 29.8 mmol) in dry toluene (10 mL) was slowly
added to the reaction mixture over a period of 40 min and stirred for 1.5 h. Later 1M
HC1 (33.5 mL) was added to the reaction mixture over a period of 20 min and stirred at
RT for 1.0 h. The two layers were separated and the aqueous layer was extracted with
toluene (2 X 15 mL). To the combined organic extracts, p-toluenesulfonic acid (0.1 g,
0.58 mmol) was added and refluxed it for 16 h. Then toluene was distilled out under
vacuum. 4.29 g of the crude product was chromatographed (20% ethyl acetate in
149IPage
H3C 25 24 CH2
BnO NaH THF
BnO CHO CH3
( EtO) 20P
Chapter III
petroleum ether) to obtain 3.37 g (77.8%) of liquid 27; by 93°C/1 mmHg [lit26 by =
98°C/1 mmHg].
3.5.2 Preparation of diethyl (2-oxohex-5-en-3-yl)phosphonate (26)
0
1. NaH, THE //'. CH3 D. (Et0) 20P
2. H2C=CH-CH 2Br P0(OEt) 2
H3C
27 26
Diethyl-2-oxopropylphosphonate 27 (3.1 g, 15.98 mmol) was added to the suspension
of sodium hydride (0.75 g, 24.0 mmol) in 10 mL THF at 0 °C. Once the anion was
formed (stirred at RT for 1.0 h), the reaction mixture was cooled to 0 °C. 1.94 g (15.98
mmol) of ally' bromide was then added to the reaction mixture and stirred at RT for 2.0
h. The reaction mixture was concentrated under vacuum and cold, saturated ammonium
chloride solution was slowly added to it. The product was extracted in diethyl ether (3 X
15 mL), washed the organic extracts with water and brine solution. The organic layer
was dried over anhydrous Na2SO4, filtered and concentrated to obtain 3.02 g of the
crude product. The product was purified using column chromatography (10% ethyl
acetate in petroleum ether) to yield 2.78 g (74.5%) of diethyl (2-oxohex-5-en-3-
yl)phosphonate as viscous liquid.
3.5.3 Preparation of (3E)-3[3-(benzyloxy)benzylidenelhex-5-en-2-one (24)
26
Phosphonate 26 (1.82 g, 7.86 mmol) was added to the suspension of sodium hydride
(0.34 g, 8.57 mmol) in 10 mL THF at -5 °C. Once the anion was formed (stirred at RT
for 1.0 h), the reaction mixture was cooled to -5 °C and aldehyde (1.5 g, 7.14 mmol) was
150IPage

Chapter III
added to it. Then stirred at RT for 1.0 h. Concentrated the reaction mixture under
vacuum and slowly added cold saturated ammonium chloride solution. The product was
extracted in diethyl ether (3 X 15 mL), washed the organic extracts with water and brine
solution. The organic layer was dried over anhydrous Na 2SO4, filtered and concentrated
to obtain 1.71 g of the crude product. The product was purified using column
chromatography (5% ethyl acetate in petroleum ether) to yield 1.36 g (65.7%) of (3E)-
3-[3-(benzyloxy)- benzylidene]hex-5-en-2-one as a light yellow viscous liquid.
3.5.4 Preparation of 8-benzy1-7-hydroxv-4-methyl-2-acetonaphthone (23a)
BnO CH3
TFA _ow
HO CH3
CH2
24
CH3
23a
5 mL of TFA and ketone 24 (0.05 g, 0.17 mmol) was refluxed for 3.0 h. Cooled the
reaction mixture to RT and added cold water to it. The acidic mixture was basified
using solid sodium bicarbonate, extracted with dichloromethane (4 X 10 mL), dried
over anhydrous Na2SO4, filtered and concentrated to obtain 0.077 g of the crude
product. The pure product obtained after column chromatography (5% ethyl acetate in
petroleum ether) was 0.029 g as a light yellow solid; mp 192 — 194 °C.
3.5.5 Preparation of 7-(benzyloxy)-4-methyl-2-acetonaphthone (23)
BnO CH3
H2SO4
BnO CH3
CH3 23
The mixture of 24 (0.05 g, 0.17 mmol), 40% sulphuric acid (2 mL), and DCM (2 mL)
was stirred at RT for 48 h. To the reaction mixture, water (5 mL) was added, which was
1511 Page

Chapter III
later neutralized with solid sodium bicarbonate and extracted it in dichloromethane (3 X
10 mL). All the organic extracts were combined, dried over anhydrous Na2SO4, filtered
and concentrated to obtain 61 mg of the crude product. The product was
chromatographed, (10% ethyl acetate in petroleum ether), to get 0.016 g of 23 as a light
yellow solid; mp 102-106 °C.
3.5.6 Preparation of (3E)-3-1 -3-(benzyloxy)benzylidenelhexane-2,5-dione (32)
BnO BnO n0
Wacker
reaction
CH2
24
32
Oxygen was passed through the mixture of PdC12 (0.14 g, 0.79 mmol), freshly prepared
CuCl (0.78 g, 7.88 mmol) in DMF (15 mL) and water (1 mL) for 1.0 h. To this mixture
2.3 g (7.88 mmol) of 24 dissolved in 7 mL of DMF was added. After stirrig and passing
oxygen at RT for 10.0 h, the reaction mixture was filtered through celite bed, washed
celite bed well with diethyl ether. To the filtrate 25 mL of water was added. The
aqueous layer was extracted with diethyl ether (7 X 20 mL). All the organic extracts
were combined, dried over anhydrous Na2SO4, filtered and concentrated to obtain 2.51
g of crude Wacker product 32. Column chromatographic purification (10% ethyl acetate
in petroleum ether) of this crude product gave 2.12 g (87.4%) of pure 32 as dark yellow
viscous oil.
3.5.7 Preparation of 7-(benzyloxy)-4-methyl-2-acetonaphthone (23)
BnO PPA
BnO CH3
CH3
23
32
152 a g e

CHO BnO
BnO
OH
CH2 36
39
BnO
Chapter III
Wacker product 32 (0.2 g, 0.65 mmol) was added to the cooled (ice-salt mixture)
polyphophoric acid (1.3 mL) and stirred for 5 min. Ice was added to the reaction
mixture and extracted it in ethyl acetate (3 X 10 mL) All the organic extracts were
combined, washed with water, dried over anhydrous Na2SO4, filtered and concentrated
to obtain 0.116 g of crude product. Pure product obtained after column chromatography
(10% ethyl acetate in petroleum ether) was 0.098 g (51.7%).
3.5.8 Preparation of carboethoxy-(a-ally1)-methylidenetriphenylphosphorane (37)
Br OEt
\ CH2
Chloroform Ph3 P
Ph
38
The mixture of stable phosphorane (10 g, 28.74 mmol) and allyl bromide (3.48 g, 28.74
mmol) in chloroform (15 mL) was refluxed for 6.0 h. The reaction mixture was
concentrated under vacuum and the salt was dissolved in water. The aqueous layer was
washed well with benzene to remove all the unreacted starting materials and basified it
using 2N NaOH solution to pH 9. The product was extracted in benzene (5 X 25 mL)
and all the organic extracts were combined. The organic extract was dried over
anhydrous Na2SO4, filtered and concentrated to obtain 7.30 g of pure allyl phosphorane
37; mp 121 — 122°C (lit32 mp 122°C).
3.5.9 Preparation of 2[3-(benzyloxy)benzylidenelpent-4-enoic acid (36)
3-benzyloxybenzaldehyde (1.6 g, 7.75 mmol), phosphorane 37 (4.0 g, 10.31 mmol) and
chloroform (15 mL) were refluxed for 24.0 h. Concentrated the reaction mixture under
153 'Pae

Chapter III
vacuum, added 10 mL of methanol and 20 N NaOH, refluxed vigorously for 1.0 h.
Removed methanol on water bath, the reaction mixture was then cooled to RT and
diluted with water. The aqueous layer was extracted with benzene (to remove all the
oxide), acidified with dilute HO to pH 2 and the product was extracted in ethyl acetate
(4 X 15 mL). All the combined organic extracts were dried over anhydrous Na 2SO4,
filtered and concentrated to obtain 0.92 g (66.2%) of acid 36 as a white solid; mp 102 —
104°C.
3.5.10 Preparation of 1-[3-(benzyloxv)phenyllpent-4-en-2-one (42)
I - + C Ph3P--\
O—CH3
1. t BuO-K-1-
eH2
Br
Ph3 P
O—CH3
tBuOK+ [Ph3P
] eH2
O-CH3 2. H 2C.._—=\
Br
Bn0 CHO BnO
CH2
42
(Methoxymethyl)triphenylphosphonium chloride (0.8 g, 2.61 mmol) was suspended in
1:1 mixture of 10 mL of THF:Toluene under nitrogen atmosphere. Potassium-t-
butoxide (KTBT) (0.29 g, 2.61 mmol) was added slowly from the round-bottom flask
through the L-shaped tube at 0 °C. At the end of the addition the reaction mixture had
become orange. After stirring for 10 min, allyl bromide (0.35 g, 2.87 mmol) in 2 mL
THF was added to it at 5 °C. The reaction mixture was stirred at RT for 1.0 h and then
cooled to 0°C. KTBT (0.26 g, 2.34 mmol) was added slowly to the reaction mixture,
which lead to its dark red coloration. The mixture was again stirred 10 min and 3-
benzyloxybenzaldehyde (0.55 g, 2.61 mmol) in 5 mL THF was added slowly at 5 °C.
The reaction mixture turned yellow from dark red. Stirred at RT for 2.0 h and quenched
it into cooled ammonium chloride solution. The product was extracted in diethyl ether
154IPage

Chapter III
(3 X 10 mL) and ethyl acetate (2 X 10 mL). The combined extract was washed with
water (2 X 10 mL) and dried over anhydrous Na2SO4. Filtration and concentration of
the filtrate gave 1.45 g of the crude product which contained desired compound,
triphenylphosphine and triphenylphosphine oxide. Separation of the product by column
chromatography (2% ethyl acetate in petroleum ether) gave 0.13 g (20.64%) of product
as yellow oil.
155 a g e

Chapter III
3.6 REFERENCES
1. Maeda, H.; Edo, K.; Ishida, N. Neocarzinostatin; Springer : Tokyo, 1997, p 287.
2. Ishida, N. J.; Miyazaki, M.; Kumagai, K.; Rikimaru, M. J. Antibiot. 1965, 18,
68.
3. (a). Napier, M. A.; Holmquist, B.; Strydom, D. J; Goldberg, I. H. Biochem.
Biophys. Res. Commun. 1979, 89, 635. (b). Koide, Y.; Ishii, F.; Hasuda, K.;
Koyama, Y.; Edo, K.; Katamine, S.; Kitame, F.; Ishida, N. J. Antibiot. 1980, 33,
342.
4. Myers, A. G. Tetrahedron Lett. 1987, 28, 4493.
5. Goldberg, I. H.; Kappen, L. S. Enediynes Antibiotics as Antitumor Agents; ed.
Border, D. D.; Doyle, T. W; Dekker, New-York, 1994, p 327.
6. Schaus, S. E.; Cavalieri, D.; Myers, A. G. Prac. Natl. Acad. Sci. USA, 2001, 98,
11075.
7. Beerman, T. A.; Poon, R.; Goldberg, I. H. Biochim. Biophys. Acta, 1977, 475,
294.
8. Kappen, L. S.; Goldberg, I. H. Nucleic Acids Res. 1985, 13, 1637.
9. Sakamoto, S.; Ogta, J.; Ikegami, K.; Maeda, H.; Cancer Treat. Rep. 1979, 62,
453.
10.Takahashi, M.; Toriyama, K.; Maeda, M.; Kikuchi, M.; Kumagai, K.; Ishida, N.
Tohoku J. Exp. Med. 1969, 98, 273.
11.Ikeda, K.; Saitoh, S.; Kobayashi, M.; Suzuki, Y.; Suzuki, F.; Tsubota, A.; Arase,
Y.; Chayama, K.; Murashima, N.; Kamada, H. J. Gastroenteral. 2000, 35, 353.
12.Berry, D. E.; Collins, J. M. Cancer Res. 1980, 40, 2405.
13.(a) Maeda, H.; Takeshita, J.; Kanamaru, R. Int. J. Pept. Protein Res. 1979, 14,
81. (b) Maeda, H.; Matsumoto, T.; Konno, T.; Iwai, K.; Ueda, M. J. Protein
Chem. 1984, 3, 181. (c) Maeda, H.; Ueda, M.; Morinaga, T.; Matsumoto, T. J.
Med. Chem. 1985, 28, 455.
14.Povirk, L. F.; Dattagupta, N.; Warf, B. C.; Goldberg, I. H. Biochemistry, 1981,
20, 4007.
156IPage

Chapter III
15.Kim, K. —H.; Kwon, B. —M.; Myers, A. G.; Rees, D. C. Science, 1993, 262,
1042.
16. (a) Hensens, 0. D.; Dewey, R. S.; Liesch, J. M.; Napier, M. A.; Reamer, R. A.;
Smith, J. L.; Schonberg, G. A.; Goldberg, I. H. Biochem. Biophys. Res.
Commun. 1983, 113, 538. (b) Edo, K.; Katamine, S.; Kitame, F.; Ishida, N.;
Koide, Y.; Kusano, G.; Nozoe, S. J. Antibiot. 1980, 33, 347.
17.Shibuya, M.; Toyooka, K.; Kubota, S. Tetrahedron Lett. 1984, 25, 1171.
18.Shishido, K.; Yamashita, A.; Hiroya, K.; Fukumoto, K.; Kametani, T.
Tetrahedron Lett. 1989, 30, 111.
19.Citterio, A.; Pesce, L.; Sebastiano, R.; Santi, R. Synthesis, 1990, 142.
20. Takahashi, K.; Suzuki, T.; Hirama, M. Tetrahedron Lett. 1992, 33, 4603.
21. Myers, A. G.; Subramanian, V.; Hammond, M. Tetrahedron Lett. 1996, 37, 587.
22. Gorth, C. F.; Rucker, M.; Eckhardt, M.; Bruckner, R. Eur. J. Org. Chem. 2000,
14, 2605.
23. Ji, N.; Rosen, B. M.; Myers, A. G. Org. Lett. 2004, 6, 4551
24. Arbuzov, A. E. J. Russ. Phys. Chem. Soc. 1906, 38, 687.
25. Kim, D. Y.; Kong, M. S.; Kim, T. H. Synth. Commun. 1996, 26, 12487.
26. Cotton, F. A.; Schunn, R. A. J. Am. Chem. Soc. 1963, 85, 2394.
27. Tilve, S. G.; Tilve, A. S.; Desai, V. G. Synth. Commun. 1996, 26, 1921.
28. Amonkar, C. P.; Patre, R. E.; Tilve, S. G. Synth. Commun. 2006, 36, 13.
29. Takas, J. M.; Jiang, X. :T. Curr. Org. Chem. 2003, 7, 369.
30. Kurangi, R.; Kinalekar, S.; Tilve, S.; Kirtany, J. Arkivoc, 2008, xii, 256.
31. Scriven, E. F. V.; Turnbull, K.; Chem. Rev. 1988, 88, 297.
32. Mali, R. S.; Tilve, S. G.; Manekar, A. R.; Yeola, S. Y. Heterocycles, 1987, 26,
121.
157 'Page