Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Zur Kenntnis der 1,3,5-Triphenylbenzole
Author(s): Uebersax, Hans
Publication Date: 1949
Permanent Link: https://doi.org/10.3929/ethz-a-000089056
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Zur Kenntnis
der 1, 3, 5-Triphenylbenzole
VON DER
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
ZUR ERLANOUNO
DER WÜRDE EINES DOKTORS DER
TECHNISCHEN WISSENSCHAFTEN
GENEHMIGTE
PROMOTIONSARBEIT
VOROELEOT VON
Hans Uebersax
Dipl. Ingenieur-Chemiker
von Oberönz (Bern)
Referent : Herr Prof. Dr. H. E. Fierz-David
Korreferent: Herr Prof. Dr.L. Blangey
v J
ZÜRICH 1949
Dissertationsdruckerei Leemann AG*.

Leer - Vide - Empty

MEINEN LIEBEN ELTERN
r
Leer - Vide - Empty

Meinen hochverehrten Lehrern,
Herrn Prof. Dr. H. E. FIERZ-DAVID
und
Herrn Prof. Dr. L. BLANGEY,
bin ich für das wohlwollende Interesse und die wert¬
vollen Anregungen, mit welchen sie diese Arbeit för¬
derten, zu bleibendem Danke verpflichtet.
Leer - Vide - Empty

Inhaltsverzeichnis
Seite
Einleitung 9
Allgemeiner Teil
I. Darstellung von Triphenylbenzol-Derivaten durch Kondensation von
Acetophenonen 10
1. Ausgangsprodukte 10
2. Kondensation der Acetophenone 12
a) Kondensation mit Kaliumpyrosulfat und Schwefelsäure .15
b) Kondensation mit Anilin und Salzsäure 17
c) Nebenprodukte der Kondensation 17
3. Tri-(aminophenyl)-benzol 19
4. Trisazo-Farbstoffe 22
1K Nitrierungsprodukte des Triphenylbenzols 23
1. Nitrierung von Triphenylbenzol 25
2. Trennung der Nitrierungsgemische nach der chromatographischen
Adsorptionsmethode 26
3. Zusammenstellung der Nitrierungsergebnisse 31
4. Konstitutionsermittlung einiger Nitrierungsprodukte des
Triphenylbenzols 31
Experimenteller Teil
I. Darstellung von Triphenylbenzol-Derivaten durch Kondensation
von Acetophenonen 35
1. Ausgangsprodukte 35
2. Kondensation von Acetophenon-Derivaten 38
a) Kondensation mit Kaliumpyrosulfat und Schwefelsäure .38
b) Kondensation in Anilin mit Salzsäure 42
c) Nebenprodukte bei der Kondensation von p-Nitroacetophenon 42
3. Tri-(aminophenyl)-benzol 44
a) Reduktion von Tri-(nitrophenyl)-benzolen 44
b) Derivate der Tri-(aminophenyl)-benzole 46
c) Ersatz der Aminogruppen 49
II. Nitrierungsprodukte des Triphenylbenzols 51
1. Nitrierung des Triphenylbenzols 51
2. Trennung der in Alkohol löslichen Nitrierungsgemische . .54
3. Konstitutionsermittlung einiger Nitrierungs-Produkte ...58
Zusammenfassung62
Literaturverzeichnis 64

Leer - Vide - Empty

Einleitung
Seit der Entdeckung des 1,3,5-Triphenylbenzols durch Engler
im Jahre 1870 bildete dieser zentrosymmetrisch gebaute aromati¬
sche Kohlenwasserstoff oft Gegenstand experimenteller wissen¬
schaftlicher Untersuchungen. So untersuchten Delacre und Mit¬
arbeiter während 30 Jahren an den Kondensationsprodukten des
Acetophenons die schrittweise Synthese des Benzolringes. Als
Endglied der zahlreichen Kondensationsprodukte trat stets das
1,3,5-Triphenylbenzol in Erscheinung. Vorländer und Mitarbeiter
betrachteten in ihren Beiträgen „zur Kenntnis der Harze und
Lacke" an Triphenylbenzol-Derivaten die Erscheinung der doppel¬
brechenden, überkühlten, amorphen Schmelzen. Aber auch auf dem
Gebiete struktureller und sterischer Untersuchungen wurde in
neuester Zeit das Triphenylbenzol herangezogen.Anderseits begegneten wir in der Literatur keiner Bemerkung
über eine Verwendung von Triphenylbenzol und Triphenylbenzol-Derivaten in der Technik oder in der angewandten Chemie.
In der vorliegenden Arbeit sollte versucht werden, aus dem
Tri-(aminophenyl)-benzol durch dreifache Diazotierung und Kupp¬
lung mit verschiedenen Komponenten der Naphthalinreihe einige
direkt ziehende Farbstoffe herzustellen. Bei der Untersuchung
eines möglichst leicht zugänglichen Ausgangsmaterials für die
Darstellung der Trisazo-Farbstoffe trat das Problem der direkten.
Nitrierung des Triphenylbenzols auf.

Allgemeiner Teil
I. Darstellung von Triphenylbenzol-Derivatendurch Kondensation von Acetophenonen
1. Au sga n gs p ro d uk te
Die Darstellung der Triphenylbenzole erfolgte ausschließlich
durch Kondensation von Acetophenonen, indem sich die Methyl-keton-Reste dreier Moleküle schrittweise unter Wasseraustritt zum
zentralen Benzolkern des Triphenylbenzols konzentrierten. Es er¬
scheint daher zweckmäßig, die meistenteils bekannten Ausgangs¬produkte kurz zu beschreiben.
Schon sehr früh fanden die Nitro-Derivate des Acetophenonsein reges Interesse, nämlich zur Zeit der Konstitutionsaufklärungdes Indigos durch Baeyer, indem Einmerüng und Engler [1]l)aus o-Nitroacetophenon Indigoblau durch Wasserabspaltung und
Reduktion mit Natronkalk und Zinkstaub synthetisierten2). Bei der
direkten Nitrierung des Acetophenons, wie sie obige Autoren
durchführten, entsteht zur Hauptsache m-Nitroacetophenon, wäh¬
rend die isomere o-Verbindung als ölige Substanz nur in kleiner
Menge auftritt, neben der spurenweise nachzuweisenden p-Ver-bindung. Zur Darstellung des p-Nitroderivates wählten wir nach
Untersuchung verschiedener Methoden die von Gevekoht [2] an¬
gegebene Acetessigester-Synthese.
p-Bromacetophenon wurde erstmals von Schweitzer [3] aus
Brombenzol und Acetylchlorid in Gegenwart von Aluminiumchlo-
!) Alle Ziffern in eckigen Klammern verweisen auf das Literaturver¬
zeichnis.
2) Eine Reproduktion dieser Indigo-Synthese gelang Gevekoht und
auch den Verfassern nicht wieder. Dagegen konnte Gevekoht aus Halogen-Derivaten des o-Nitroacetophenons durch Kochen in alkoholischer Ammo¬
niumsulfidlösung Indigo erhalten.

— 11 —
rid und Schwefelkohlenstoff synthetisiert, während über tn-Brom-
acetophenon in der Literatur keine Angaben aufzufinden waren.
Zur Synthese dieser Verbindung sind viele Wege gangbar; wir
wählten die Darstellung über das leicht zugängliche m-Nitroaceto-
phenon, das nach der Reduktion zum Amin mit der bekannten Me¬
thode von Sandmeyer einen Ersatz der Aminogruppe durch Brom
gestattet.
m-Nitroacetophenon stellte man nach der von Rupe [4]angegebenen Methode durch Nitrieren mit Mischsäure dar. Die
Nitrierung in m-Stellung verläuft bei Einhaltung tiefer Reaktions¬
temperaturen sehr glatt. Es ist jedoch zu erwähnen, daß beim
Lösen des Acetophenons in der Schwefelsäure (d -= 1,84) die
Lösungstemperatur nicht über 10° ansteigen darf, da sich sonst,
wie sich zeigte, zwei Acetophenon-Moleküle unter Wasseraustritt
zu Dypnon und Dypnon-Derivaten kondensieren, was schließlich
zu einem dunklen, öligen Endprodukt führt.
p-Nitroacetophenon war durch direkte Nitrierungnicht zugänglich. Die bekannten Methoden, die zu dem gewünsch¬ten Produkte führen, sind sehr weitläufig und mit schlechten Aus¬
beuten verbunden. Die besten Resultate waren mit der von Geve-
koht angegebenen Methode zu erzielen: Ausgehend von p-Nitro-
benzoylchlorid wird mit Natriumacetatessigester zunächst p-Nitro-
benzoylacetessigester hergestellt. Der substituierte Acetessigesterwird als ein Öl erhalten und erstarrt erst bei Kühlung unter 10°,
während die reine Verbindung, die besser nach Biilow und
Hailer [5] mit Acetessigester und Natriumalkoholat erhältlich ist,
bei 53° schmilzt. Die Verseifung des substituierten Acetessigesters
gelingt am besten mit 30proz. Schwefelsäure, wobei neben dem
gewünschten p-Nitroacetophenon in beträchtlicher Menge p-Nitro-benzoesäure entsteht, welche durch Waschen der ätherischen Lö¬
sung mit Bikarbonat vom Nitroacetophenon zu trennen ist. Es hatte
sich gezeigt, daß die Ausbeuten nach der Methode von Gevekoht
über ein „unreines" Rohprodukt besser sind als die Ausbeuten
über den reinen, nach Biilow hergestellten Acetessigester.
Versuche, p-Nitroacetophenon nach Grignard mit Methyl-
magnesiumbromid und Nitrobenzoylchlorid in Analogie zu einer
neueren Acetophenon-Synthese darzustellen, schlugen fehl [6].

- 12 —
p-Bromacetophenon wird nach den Literaturangabenmeist nach der Friedel-Krafts-Reaktion über Brombenzol mit
Essigsäureanhydrid und Aluminiumchlorid in Schwefelkohlenstoff
als Lösungsmittel dargestellt [7, 8]. Wir benützten eine an Grog¬gins [9] angelehnte Darstellungsweise (für p-Chloracetophenonausgearbeitet), die den unangenehmen Schwefelkohlenstoff durch
einen Brombenzol-Überschuß als Lösungsmittel ersetzt. Die Va¬
kuumdestillation im Fraktionierkolben gestattet eine gute Tren¬
nung vom Lösungsmittel. In Alkohol umkristallisiert, erhielt man
weiße Blättchen von p-Bromacetophenon vom Schmelzpunkt 51°,
in Übereinstimmung zu den Angaben von Schweitzer.
Angaben zur Darstellung von m-Bromacetophenonließen sich, wie erwähnt, keine finden. Zur Synthese dieses Pro¬
duktes diente der folgende Weg: Aus m-Nitroacetophenon erhält
man durch Reduktion nach Bêchamp und Brimmeyer [10] das m-
Aminoacetophenon, das schon von Engler [11] beschrieben wor¬
den ist. Das Amin ist in sehr verdünntem Alkohol löslich und läßt
sich gut aus dem alkalischen Eisenschlamm extrahieren. Es löst
sich in 0,5-n Bromwasserstofflösung und läßt sich mit 1-n Natrium¬
nitritlösung nach den üblichen Methoden diazotieren. Der Ersatz
der Diazoniumgruppe durch Brom erfolgt mit Kupferbromür und
Bromwasserstoff in heißer Lösung nach der Methode von Sand¬
meyer [12]. Durch Extraktion mit Äther und Destillation im
Vakuum resultiert ein hellgelbes Öl, das bei Zimmertemperaturflüssig bleibt. Erst in der Kälte erstarrt das erhaltene m-Brom¬
acetophenon und schmilzt wiederum bei 7°.
2. Kondensation der Acetophenone
Seit der Entdeckung der Kondensation des Acetophenonsunter Einwirkung von wasserabspaltenden Agentien zu Triphenyl-benzol durch Engler [13] bildete das Acetophenon als einfachstes
Arylalkylketon öfters Gegenstand wissenschaftlicher Untersuchun¬
gen. Während vielen Jahren erforschten Delacre und Mitar¬
beiter [14] die schrittweise Synthese des Benzolringes vor allem
an Acetophenon-Abkömmlingen. Die Phenylgruppe dieses ge¬
mischten Ketons war für diese Untersuchungen von Vorteil, weil

— 13 -
dadurch die Kondensation begünstigt und vor allem aber in über¬
sichtlichere Bahnen geleitet werden konnte. Entsprechende Kon¬
densationen bei Aceton führten zu zahlreichen, unübersichtlichen
Kondensations- und Polymerisations-Produkten. So hatte Delacre
im Laufe von 30 Jahren neben dem ersten Kondensationsprodukt,dem Dypnon3), durch Kondensation und Polymerisation mit ge¬
eigneten Agentien aus Acetophenon u. a. vier isomere Pinakone,
neun isomere Pinakoline, fünf isomere Pinakonalkohole und fünf
isomere Kohlenwasserstoffe C25H22 isoliert und beschrieben. Alle
diese Produkte geben durch teilweise pyrogene Zersetzung bei der
Destillation stets Triphenylbenzol, was die Stabilität des Benzol¬
sechsringes erweisen sollte.
Die Vielzahl dieser Zwischenprodukte läßt vermuten, daß die
Synthese des Triphenylbenzols nicht durch einfache Kondensation
von 3 Molen Acetophenon zu erklären ist, sondern daß verschie¬
dene Zwischenstufen zu berücksichtigen sind.
Aber auch zur Aufklärung der Wirkungsweise von Kondensa¬
tionsmitteln fand Acetophenon häufig Anwendung. Nach den
ersten Untersuchungen von Engler mit Phosphorpentoxyd, Salz¬
säure und Zinkchlorid als wasserentziehende Mittel berichten zahl¬
reiche weitere Verfasser über die Einwirkung von Kondensations¬
mitteln auf Acetophenon oder auf Arylalkylketone [15—24]. Odell
und fiines [25] prüften die Pyrosulfate des Natriums und Kaliums
als Kondensationsmittel für Ketone und erzielten damit sehr gute
Ergebnisse, so daß wir nach unten angeführten Vorversuchen eben¬
falls zu diesem Kondensationsmittel griffen.Arbeiten über die Kondensation von Acetophenon-Derivaten
zu Triarylbenzolen lassen sich dagegen in der Literatur nur ganz
wenige auffinden. Auch begegnete man keinem Derivat, das in
der Technik Interesse oder Verwendung gefunden hätte. Alle
Publikationen galten der experimentellen Erforschung des Kon¬
densationsvorganges oder der speziellen symmetrischen Struktur
dieser Produkte. Über die Kondensation substituierter aromatischer
Methylketone gibt einzig eine Arbeit von Bernhauer [26] Auskunft,
die den Einfluß der Substituenten auf die Kondensationsfähigkeit
s) Ein dem Mesityloxyd entsprechendes Kondensationsprodukt.

— 14 —
des Ketons untersucht. Bei Verwendung von Kaliumpyrosulfat undSchwefelsäure als wasserentziehende Mittel war eine Gesetz¬
mäßigkeit zu erkennen in dem Sinne, daß vor allem dann Kon¬
densation unter Bildung von Triarylbenzolen stattfindet, wenn
negative oder keine Substituenten im aromatischen Kern vorhan¬den sind, während positive Substituenten den Kondensationsvor¬
gang verhindern.
Die Kondensation der Acetophenone erfolgt stufenweise, in¬
dem sich zunächst zwei Moleküle Acetophenon unter Wasser¬
abspaltung zu Dypnon, einem dem Mesityloxyd analogen Produkt,verbinden. Dieses Zwischenprodukt ist bei milderen Kondensa¬
tionsbedingungen gut zu fassen. Den Kondensationsvorgang in
zweiter Stufe kennen wir nicht genau. Es gelang uns nicht, höher
kondensierte oder polymerisierte Zwischenstufen zu fassen, wie
sie auf anderem Wege erhältlich sind [14]. Aus der Beobachtung,daß bei der Selbstkondensation des reinen Dypnons Triphenyl-benzol in viel kleinerer Menge entsteht als bei der Kondensation
des Dypnons bei Anwesenheit von Acetophenon, können wir an¬
nehmen, daß sich zur Hauptsache ein weiteres Acetophenon-Mole-kül an Dypnon anlagert und unter Abspaltung von 2 Molen Wasser
Triphenylbenzol bildet. Wir haben gezeigt, daß aus reinem Dinitro-
dypnon durch Selbstkondensation mit K2S207 und konz. Schwefel¬
säure Trinitrophenyl-benzol erhalten werden kann, jedoch in ge¬
ringerer Ausbeute als bei Anwesenheit von p-Nitroacetophenon.Bei der Selbstkondensation des Dinitrodypnons verläuft die Kon¬
densation über andere Zwischenstufen, die sich ohne teilweise
Spaltung nicht erklären lassen. — Versuche, aus Dypnon und p-
Nitroacetophenon, ein Mononitrotriphenylbenzol zu synthetisieren,bestätigen obige Beobachtungen, indem aus der Kondensations¬masse Triphenylbenzol neben Tri-nitrophenyl-benzol zu isolieren
waren, während das gesuchte p-Mononitrotriphenylbenzol nicht
aufzufinden war.
Die Wasserabspaltung erfolgt bei genügend hoher Tempera¬tur auch ohne wasserentziehende Agentien, wie Engler und
Dengler [27] gezeigt haben. Wasserentziehende Agentien beschleu¬
nigen jedoch in bekannter Weise die Kondensation und gestatteneinen guten Verlauf bei niedrigen Temperaturen. Um die Konden-

— 15 —
sation in die gewünschte Richtung zu lenken, dürfen nur saure
Agentien zur Anwendung gelangen. (Alkalien beeinflussen die
Kondensation in anderer Richtung und scheinen eher eine Poly¬merisation zu begünstigen.) In siedendem Anilin mit einer Spur
von Salzsäure gelingt die Kondensation von Acetophenon zu Tri-
phenylbenzol glatt, während Anilin allein zu ganz anderen Pro¬
dukten führt. Ebenso verläuft die Kondensation in Pyrosulfat und
wenig konz. Schwefelsäure gut, mit jeder der Komponenten für
sich allein jedoch nicht4).
CH,H.SO,
K2S..O, ^ "' KjS^ + HjSO,"^
K,S20_,H.SO,
(R = N02 bzw. Br)
a) Kondensation mit einem Molekül Acetophenon (subst.)
b) Selbstkondensation des subst. Dypnons.
a) Kondensation mit Kaliumpyrosulfat und Schwefelsäure
Kaliumpyrosulfat erwies sich, wie oben erwähnt, als gutes
Kondensationsmittel für Acetophenon-Derivate. In Übereinstim¬
mung mit Bernhauer [26] fanden wir, daß die Kondensation von
der Art des Substituenten im Benzolkern abhängig ist, und zwar
begünstigt der Substituent die Kondensation mit zunehmend nega¬
tivem Charakter. Es gelang nicht, Aminoacetophenon oder N-Ace-
tylaminoacetophenon zu entsprechenden Triphenylbenzolen zu
kondensieren. Der erwähnten Gesetzmäßigkeit entsprechend war
4) Bei Aceton dagegen ist die Kondensation mit konz. Schwefelsäure
eine geläufige Darstellungsweise.

- 16 —
von Nitroacetophenon ein günstigeres Kondensationsergebnis zu
erwarten als von den Halogenacetophenonen, was auch mit den
praktischen Erfahrungen übereinstimmte. (Bei p-Nitroacetophenon
beeinträchtigt die schwierige Aufarbeitung die Ausbeute an reinem
Tri-(nitrophenyl)-benzoL) Der Einfluß des negativen Substituen-
ten ist in meta am größten und nimmt nach para ab. Aus o-Nitro-
acetophenon konnte kein Tri-(o-nitrophenyl)-benzol synthetisiertwerden. Diese Erscheinung ist damit zu erklären, daß der Sub¬
stituent in ortho die Synthese des Benzolkernes sterisch nicht
gestattet.
Im Gegensatz zu Odell und Hines war bei der von ihnen an¬
gegebenen Temperatur von 45° auch nach 52 Stunden Reaktions¬
dauer, kein Triphenylbenzol erhältlich. Im Verlaufe der Unter¬
suchungen erwies sich für die Kondensation eine während 5 Stun¬
den von 50—85° gesteigerte, gleichmäßige Erwärmung am erfolg¬reichsten. Zur Erzielung optimaler Resultate war eine Reaktions¬
dauer von 17 Stunden bei Nitroacetophenon und von 8 Stunden bei
Bromacetophenon notwendig.Die Kondensation ist stets in erheblichem Maße von Neben¬
reaktionen begleitet. Auch lassen sich aus der Kondensationsmasse,
wie wir gezeigt haben, Zwischenprodukte vom Dypnontypus iso¬
lieren. Le Fèvre [28] gelang es, aus dem Reaktionsgemisch bei der
Triphenylbenzol-Synthese 2,4,6-Triphenylpyrylium zu isolieren,
das mit einem früher von ihm dargestellten Produkte identisch war.
Die Aufarbeitung der Tri-(nitrophenyl)-benzole bot einige
Schwierigkeiten, da sich die begleitenden harzigen Nebenproduktenur schwer trennen ließen. Zudem sind die Tri-(nitrophenyl)-ben-zole sehr schwer löslich. Das p-Isomere löst sich in o-Dichlor-
benzol spurenweise und in siedendem a-Chlornaphthalin nur sehr
wenig. (In diesen Lösungsmitteln lösen sich die Verunreinigungenebenfalls und fallen teilweise mit dem Produkt wieder aus.) Durch
Behandlung mit Aktivkohle war schließlich ein reines Produkt er¬
hältlich, doch nur unter erheblichen Verlusten. Die Lösung mußte
heiß bei 2*50° filtriert werden, denn bei der geringsten Abkühlungkristallisierte die Substanz in kleinen, gebogenen Nadeln rasch aus.
Die eingehendere Beschreibung der Triphenylbenzole ist aus dem
experimentellen Teil ersichtlich.

— 17 —
b) /Condensation mit Anilin und Salzsäure
Reddelien [29] fand in seinen Studien über die Selbstkonden¬
sation bei Anilen, daß Acetophenonanil sich unter Einwirkung von
wenig Salzsäure bei 160° katalytisch zu Dypnonanil und in zweiter
Stufe zu Triphenylbenzol kondensiert unter Austritt von 3 Mole¬
külen Anilin. Diese Anilkondensation ist ein Analogon zur Selbst¬
kondensation des Acetophenons, nur daß an Stelle von Wasser
Anilin austritt. Mit dieser Beobachtung hatte Reddelien eine ein¬
fache Kondensationsmethode für Acetophenone gefunden. In einer
spateren Notiz [30] bemerkte derselbe Verfasser, daß in Anilin
und wenig Salzsäure mit guter Ausbeute zahlreiche Acetophenon-Derivate zur Kondensation gebracht werden können. Allerdingswurde nur das mit besonders ausgeprägten kristallinen Eigen¬schaften ausgezeichnete l,3,5-Tri-(biphenyl)-benzol erwähnt. In
verschiedenen Versuchen gelang es uns aber nicht, mit dieser Me¬
thode Nitroacetophenone oder Aminoacetophenone (auch N-Sub-
stituierte) zur Kondensation zu bringen, selbst bei Temperaturenüber 200°. Hingegen leistete diese Methode bei der Darstellung
\on Triphenylbenzol ausgezeichnete Dienste, indem schon nach
30 Minuten Reaktionsdauer bei 180° Ausbeuten an Triphenylben¬zol bis zu 65 o/o zu erreichen waren.
c) Nebenprodukte bei der Condensation
Wie an anderem Orte erwähnt, ist die Kondensation stets von
Nebenreaktionen begleitet, die teilweise zu Polymerisaten führen,
während anderseits die Kondensation teilweise nur bis zur Zwi¬
schenstufe erfolgt. Es konnten daher im besten Falle höchstens
65 u'o der theoretisch möglichen Ausbeute erhalten werden. Die
Begleitprodukte sind zur Hauptsache in Alkohol löslich und lassen
sich damit extrahieren. In der Kälte scheiden sie sich als braune,
harzige Masse ab. Wir befaßten uns daher etwas näher mit den
im Alkoholextrakt löslichen Produkten aus der p-Nitroacetophe-
non-Kondensation, um den Hauptbestandteil daraus zu isolieren.
Versuche, durch Extraktion oder Kristallisation von den Harzen zu
trennen, waren erfolglos. Erst die Anwendung der chromatographi-

— 18 —
sehen Adsorptionsmethode auf diese Gemische gestattete eine
saubere Trennung von den harzigen Bestandteilen und eine genaue
Beurteilung des Gemisches.
Das getrocknete, pulverisierte Gemisch löste sich in Petrol-
äther und wurde mit diesem Lösungsmittel auf der Adsorptions¬säule (als Adsorbens diente ein Aluminiumoxyd von der Aktivität
1—2) aufgezogen. Eine hellgelbe Zone setzte sich über die ganze
Säule fest, während zu oberst in schmalen Ringen die harzigen
Polymerisate adsorbiert wurden. Die breite, hellgelbe Zone er¬
schien völlig einheitlich und konnte mit Petroläther-Benzol-Ge-
mischen eluiert werden. Die vereinigten Petroläther-Benzol-Eluate
ergaben ein gut kristallisierendes, hellgelbes Produkt, das 73 o0
des Gemisches ausmachte. Aus den mit Äther-Benzol eluierten har¬
zigen Bestandteilen war keine einheitliche Substanz zu isolieren,so daß wir uns auf die Identifizierung des kristallinen Produktes
beschränkten. Die isolierte Substanz ist nicht säure-alkaliempfind-lich und fluoresziert auch nicht in alkoholischer Lösung. Das
Fehlen dieser Eigenschaften ließ die Vermutung auf ein Pyrylium-
Derivat, wie es von Le Fèvre aufgefunden wurde, unwahrschein¬
lich erscheinen. Die eluierte Substanz kann jedoch für sich allein
oder in Gegenwart von p-Nitroacetophenon weiter zum Triphenyl-benzol kondensiert werden, womit anzunehmen ist, daß es sich
um ein Zwischenprodukt der Kondensation handeln muß. Da die
Substanz überdies als einheitliches Produkt ohne isomere Verbin¬
dung im Chromatogramm vorhanden war, vermuteten wir zunächst,daß es sich um ein Dinitrodypnon handelte. Sollte diese hypo¬thetische Annahme stimmen, dann müßte auch die vorhandene
Ketogruppe in Erscheinung treten und nachzuweisen sein. Die
Analysen-Resultate, die mit den berechneten Werten übereinstimm¬
ten, genügten noch nicht zur genauen Identifizierung der Substanz.
Es gelang jedoch, ein gut kristallisierendes Semicarbazon der Ver¬
bindung herzustellen, womit die Anwesenheit einer Ketogruppeerwiesen sein dürfte.
p-p'-Dinitrodypnon ist durch Selbstkondensation von 2 Mole¬
külen Acetophenon und Austritt von 1 Molekül Wasser entstanden
und stellt somit ein faßbares Zwischenprodukt bei der Kondensa¬
tion von Nitroacetophenon zu Tri-(nitrophenyl)-benzol dar.
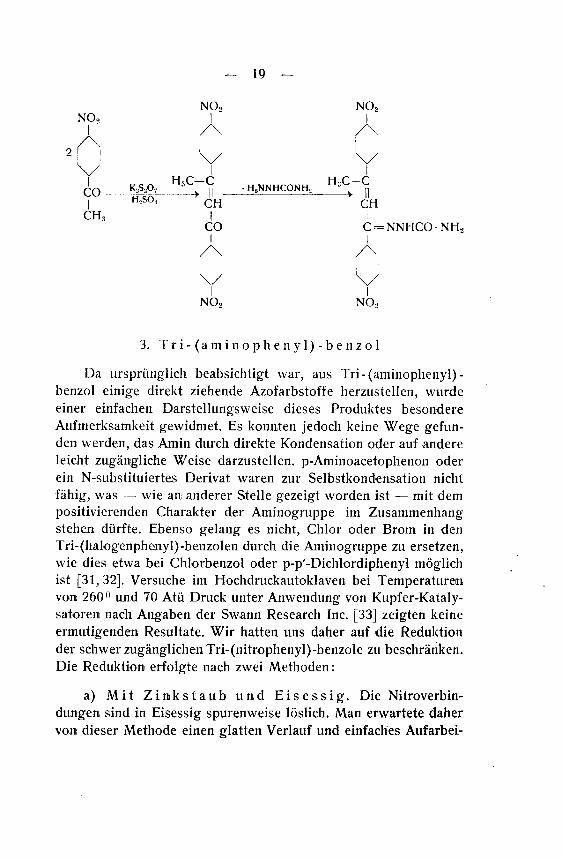
- 19 —
NO,
NO,
K,S20,
H=SO;
CH
13C-C— II —
CHi
H2NNHCONH2H3C -C
IICH
1
1
CO
1
/\
C = NNHCONH;1
01
cNO, NO,
3. Tri-(aminophenyl)-benzol
Da ursprünglich beabsichtigt war, aus Tri-(aminophenyl)-benzol einige direkt ziehende Azofarbstoffe herzustellen, wurde
einer einfachen Darstellungsweise dieses Produktes besondere
Aufmerksamkeit gewidmet. Es konnten jedoch keine Wege gefun¬den werden, das Amin durch direkte Kondensation oder auf andere
leicht zugängliche Weise darzustellen. p-Aminoacetophenon oder
ein N-substituiertes Derivat waren zur Selbstkondensation nicht
fähig, was — wie an anderer Stelle gezeigt worden ist — mit dem
positivierenden Charakter der Aminogruppe im Zusammenhangstehen dürfte. Ebenso gelang es nicht, Chlor oder Brom in den
Tri-(halogenphenyl)-benzolen durch die Aminogruppe zu ersetzen,
wie dies etwa bei Chlorbenzol oder p-p'-Dichlordiphenyl möglichist [31,32]. Versuche im Hochdruckautoklaven bei Temperaturenvon 260° und 70 Atü Druck unter Anwendung von Kupfer-Kataly¬satoren nach Angaben der Swann Research Inc. [33] zeigten keine
ermutigenden Resultate. Wir hatten uns daher auf die Reduktion
der schwer zugänglichen Tri-(nitrophenyl)-benzole zu beschränken.
Die Reduktion erfolgte nach zwei Methoden:
a) Mit Zinikstaub und Eisessig. Die Nitroverbin¬
dungen sind in Eisessig spurenweise löslich. Man erwartete daher
von dieser Methode einen glatten Verlauf und einfaches Aufarbei-

— 20 —
ten des schwer dampffluchtigen Amins. Nach 8stündiger Reak¬
tionsdauer zeigte sich aber, daß in der Reduktionslösung kein Amin
vorlag, während anderseits auch keine Nitroverbindung mehr zu
erkennen war. Titrationsversuche mit Natriumnitrit deuteten auf
völlige Abwesenheit von primären Aminen. Während der Reduk¬
tion in heißer Eisessiglösung wurde das Amin acetyliert, und man
erhielt ein nicht kristallisierbares N-Acetylgemisch. Erst nach dem
Verseifen mit 20proz. alkoholischer Kalilauge fiel das Amin in
feinen Nädelchen aus.
b) Reduktion mit Zinnchlorur und Salzsäure.
Um das schwer dampfflüchtige Amin möglichst in einfacher Weise
zu erhalten, wählte man die Reduktion mit Zinnchlorur, die über
das Zinndoppelsalz zum Amin führt. (Zinnchlorürlösung ist schon
in der Kälte ein sehr wirksames Reduktionsmittel.) Nach zweistün¬
digem Erhitzen unter Einleitung von trockenem Salzsäuregas kri¬
stallisierte beim Abkühlen das Zinndoppelsalz des Amins aus. Das
in wenig Wasser lösliche Doppelsalz wurde stark alkalisch ge¬
macht, worauf das Amin in Flocken ausfiel. Das Amin wurde über
das in stark konz. Salzsäure unlösliche Chlorhydrat gereinigt. Zur
Reindarstellung war jedoch noch eine Filtration über Aluminium¬
oxyd mit Essigester erforderlich.
Das Amin scheint an der Luft ziemlich beständig zu sein; nach
48 Stunden waren äußerlich keine Veränderungen zu bemerken.
Erst nach mehrtägigem Stehen nahm das Amin dunklere Farbe an.
Das Tri-(p-aminophenyl)-benzol ist ziemlich stark basisch und löst
sich schon in sehr verdünnter Säure. Die weiteren Eigenschaftender Amine sind im experimentellen Teil angeführt.
Zur Charakterisierung dieser noch nicht beschriebenen Amine
haben wir einige Derivate hergestellt. Die Derivate zeichnen sich
teilweise durch schlechte Kristallisationsfähigkeit aus, was der
verzweigten Struktur des Moleküls zuzuschreiben ist, so daß die
Aufarbeitung der Derivate einige Schwierigkeiten verursachte.
• Benzyliden-Derivate: Gibt man zu den in Alkohol
gelösten Aminen p-Nitrobenzaldehyd in kleinem Überschuß, so
fällt nach kurzem Erwärmen das rötliche, in Alkohol unlösliche
Benzyliden-Derivat aus, das aus Chlorbenzol umkristallisiert wird.

— 21 —
Toluolsulfamid-Derivate: Dieses Derivat wurde
nicht nach den üblichen Methoden hergestellt [34], da dasselbe in
Natronlauge fast unlöslich ist. Die Amine wurden in Aceton ge¬
löst, mit wenig Dimethylanilin versetzt, worauf das Toluolsulfo-
säurechlorid in die Lösung zugegeben wurde. Die Umkristallisa-
tion der weißen Toluolsulfamid-Verbindungen erfolgte in ver¬
dünnter alkoholischer Lösung.Die Benzoyl-Derivate würden auf gleiche Weise dar¬
gestellt. Bemerkenswerterweise liegt der Schmelzpunkt der p-Ver-
bindung unter demjenigen der isomeren m-Verbindung.
Die Darstellung der Acetyl-Derivate nach den bekann¬
ten Methoden war mit Schwierigkeiten verbunden. Eine reine Tri-
acetyl-Verbindung war überhaupt nicht zugänglich, weil stets eine
oder mehrere Aminogruppen höher acetyliert wurden. Durch ener¬
gische Acetylierung mit Acetanhydrid bei 100° wurde ein kristal¬
lines Produkt erhalten, doch zeigen die Analysen zu hohe N-Werte,
was auf eine teilweise unvollständige Hexa-Acetylierung schließen
läßt.
Ersatz der Amino-Gruppen
Tri-(aminophenyl)-benzol ist in verdünnter Salzsäure gut lös¬
lich und läßt sich überraschenderweise sehr leicht dreifach diazo-
tieren. Zum Konstitutionsbeweis ersetzten wir die Amino-Gruppen
durch Brom und stellten die Identität mit den durch direkte Kon¬
densation erhaltenen Tri-(bromphenyl)-benzolen fest. Die Tat¬
sache, daß die Azide zur Charakterisierung der Amine brauchbare
Dienste leisten, kann durch die gemachten Beobachtungen bestä¬
tigt werden. Die leichte Darstellungsweise und die hohe Reinheit
der Rohprodukte verdienen in Übereinstimmung mit Feststellun¬
gen von Dübendorfer [35] erwähnt zu werden.
Die Diazotierung der Aminogruppen erfolgte nach den be¬
kannten Methoden mit eingestellter Natriumnitrit-Lösung, womit
gleichzeitig die Amino-Gruppen im Molekül bestimmt werden
konnten. Der Titrationsverlauf zeigt, daß sich alle drei Amino-
Gruppen gleichzeitig, ohne gegenseitige Behinderung diazotieren
lassen. Es waren keine Zwischenstufen der Diazotierung zu er¬
kennen, ein Hinweis darauf, daß die Amino-Gruppen an den

— 22 —
Phenylresten sich gegenseitig wenig beeinflussen und eher „ex¬
ternen" Charakter haben. Mit der Methode von Sandmeyer gelanges, auf die früher beschriebene Weise die Aminogruppen durch
Brom zu ersetzen. Schwierigkeiten bot die Aufarbeitung der in
kleiner Ausbeute entstandenen Bromverbindung, die erst nach
mehrmaligem Umkristallisieren aus Essigester und nach Behand¬
lung mit Aktivkohle analysenrein erhältlich war. Durch diese Ope¬ration konnte die Identität mit den durch Kondensation aus Brom-
acetophenon erhaltenen Produkten festgestellt werden.
Die sehr leicht zugänglichen Azide wurden durch dreifaches
Diazotieren der Amine und Versetzen der kongosauren Diazonium-
Lösung mit Natriumazid-Lösung hergestellt. Mit der sogleich ein¬
setzenden Stickstoffentwicklung schieden sich die Azide mit fast
weißer Farbe in körnigen Flocken aus. Nach 15 Minuten konnte
filtriert werden. Keine der dargestellten Amino-Derivate erreichten
die hervorragende Kristallisationsfähigkeit der Azide, die schon
nach einmaligem Lösen in wenig erwärmtem Essigester in Nadeln
auskristallisierten; überdies war die Ausbeute fast quantitativ.Dagegen sind die Azide äußerst lichtempfindlich und ver¬
färben sich unter Belichtung rasch von gelb nach braun. Mit konz.
Schwefelsaure tritt sofort die charakteristische Zersetzung unter
Stickstoffentwicklung ein, was an den aufsteigenden Bläschen zu
erkennen ist. Auf Zusatz von Schwefelsäure findet keine Farbreak-
tion statt. Gegen Schlag scheinen die Azide unempfindlich zu sein :
durch indirektes Erhitzen verpuffen die Azide mit gelber Flamme.
Oberhalb des Schmelzpunktes zersetzen sie sich langsam. Die
Stickstoff-Analysen erweisen die große Reinheit der nur einmal
aus Essigester umkristallisierten Verbindungen.
4. Trisazo-Farbstoffe aus Tri-(aminophenyl)-benzol
Es war beabsichtigt, aus den Tri-(aminophenyl)-benzoleneinige, wenn möglich direkt ziehende Trisazo-Farbstoffe herzu¬
stellen. Da die dreifache Diazotierung der Triamine ohne die ge¬
ringsten Schwierigkeiten erfolgte, schien einer einfachen Darstel-
lumgsweise der Farbstoffe nichts im Wege zu stehen.

- 23 -
Die Trisazo-Farbstoffe wurden durch Kupplung der dreifach
diazotierten Amine mit folgenden Naphthalin-Derivaten herge¬
stellt: Acetylaminonaphtholdisulfosäure 1,8,3,6 (Acetyl H-Säure),
Naphtholdisulfosäure 2, 6, 8 (Q-Säure), Aminonaphtholsulfosäure
2, 5, 7 (J-Säure) und Phenyl-J-Säure. Die Kupplung in sodaalkali¬
scher Lösung verlief ohne Schwierigkeiten.Da die Nuancen der erhaltenen Farbstoffe von orange nach rot
variierten, wurde ein Vergleich mit entsprechenden Monoazo-Farb-
stoffen (durch Kuppeln von diazotiertem Anilin mit den erwähnten
Komponenten erhalten) angestellt. Tatsächlich war zwischen der
3proz. Ausfärbung des Orange G [47] und dem entsprechendenTrisazo-Farbstoff aus Tri-(m-aminophenyl)-benzol und G-Säure,
kaum ein Unterschied zu sehen, während der isomere p-Farbstoffeine Farbvertiefung nach rot aufwies. Analog waren die Verhält¬
nisse bei den übrigen Farbstoffen mit den aufgezählten Kompo¬
nenten.
Die Tatsache, daß die erhaltenen Trisazo-Farbstoffe die glei¬
chen Nuancen wie die entsprechenden Monoazo-Farbstoffe auf¬
weisen, dürfte eine Folge des teilweisen Unterbruches der Kon¬
jugation der Kohlenstoffdoppelbindungen zwischen den einzelnen
Phenylresten sein.
Trotz der erheblichen Molekülgröße der Farbstoffe ist prak¬
tisch keine Affinität zu Baumwolle vorhanden. Einzig die J-Säure-
Farbstoffe ziehen erwartungsgemäß am besten auf Baumwolle auf,
doch ist auch bei diesen Farbstoffen die Affinität ungenügend. Die
unbefriedigenden Eigenschaften der erhaltenen Trisazo-Farbstoffe
gaben Anlaß, keine weiteren Versuche zu unternehmen.
II. Nitrierungsprodukte des Triphenylbenzols
Um der unbefriedigenden Darstellung der Tri-(nitrophenyl)-benzole durch Kondensation aus Nitroacetophenon eine bessere
Darstellungsmethode entgegenzusetzen, sollte versucht werden,
die gewünschten Produkte durch Nitrierung des leichter zugäng¬
lichen Triphenylbenzols zu erhalten. Die Entdecker des Triphenyl¬
benzols versuchten schon die Nitrierung und wollten durch Be-

— 24 —
handeln des Triphenylbenzols mit rauchender Salpetersäure eine
Trinitro-Verbindung erhalten haben [36]. In einem weiteren Bericht
kündigten Engler und Bertold [37] weitere Versuche über die Ni¬
trierung an, doch waren keine fortsetzenden Berichte zu diesem
Thema mehr gefolgt. Erst Meilin [38] stellte neue Versuche über
die Nitrierung des Triphenylbenzols an, die er in Eisessiglösungdurchführte. Dabei hatte er zwei isomere Verbindungen erhalten:
ein in Eisessig unlösliches Produkt, das er als at-Tetranitrotriphe-nylbenzol bezeichnete, und eine in Eisessig lösliche /3-Tetranitro-
Verbindung. (Es sei vorweg genommen, daß es sich bei dieser
„Verbindung" um ein Gemisch von mehreren Nitro-Verbindungen
handelt.) Die nächste Publikation über die Nitrierung von Triphe-nylbenzol erfolgte 40 Jahre später von Vorländer und Mitarbeitern
L3Q]. Diese Autoren erhielten bei „milder" Nitrierung in Eisessigeine gut kristallisierende Mononitro-Verbindung, die sie durch
Abbaureaktionen als p-Mononitro-triphenylbenzol bezeichneten.
Daneben bestätigten sie das bisher bekannte «,-Tetranitro-Produkt,während sie aus den in Eisessig löslichen Produkten drei verschie¬
dene amorphe Gemische, die nicht näher untersucht wurden, neben
dem von Meilin beschriebenen /S-Tetranitrotriphenylbenzol iso¬
lieren konnten.
Auf Grund der bisher in der Literatur vermerkten Beobach¬
tungen erschien es uns nicht ausgeschlossen, unter geeigneten Be¬
dingungen eine Tri-(nitrophenyl)-benzol-Verbindung mit den
Nitrogruppen in den Phenylresten zu erhalten. Doch deuteten die
Ergebnisse der angestellten Versuche auf einen anderen Verlauf
der Nitrierung, der die Bildung der gewünschten Trinitro-Verbin¬
dung nicht gestattet.Aus den Versuchen Vorländers ist ersichtlich, daß die Nitrie¬
rung des Triphenylbenzols nicht einfach verläuft und jedenfallsmehr als nur zwei isomere Verbindungen zu erwarten sind. Be¬
rücksichtigt man die zentrosymmetrisch verzweigte Struktur des
Triphenylbenzols, so erkennt man sofort die unabsehbare Zahl der
theoretisch möglichen Isomeren. Für nur zwei Nitrogruppen im
Molekül ergeben sich mindestens 16 isomer mögliche Verbindun¬
gen, unter der Einschränkung, daß die Nitrogruppen nie in ortho-
Stellung zueinander stehen und die Phenylreste frei drehbar sind.

— 25 —
Bei der Nitrierung treten nicht nur zwei Substituenten ein, son¬
dern, wie wir gezeigt haben, sind von der Mononitro-Verbindung
bis zur Hexanitro-Verbindung alle Nitrierungsstufen nachzuweisen,
so daß die Schar der theoretisch möglichen Isomeren noch weit
höher zu stehen kommt. Die Zahl der isomeren Verbindungen wird
jedoch durch eine experimentell erkannte Gesetzmäßigkeit etwas
begrenzt, indem bei jeder Nitrierung die erste Nitrogruppe in den
zentralen Kern eintritt. Es war bei keiner der zahlreichen Nitrie¬
rungen möglich, eine Verbindung, die nur Nitrogruppen in den
Phenylresten enthielte, zu isolieren. Eine solche Verbindung wäre
an der Schwerlöslichkeit sofort zu erkennen und hätte in Analogie
zu den beschriebenen Kondensationsprodukten identifiziert werden
können. Die erwähnte Gesetzmäßigkeit wurde durch zahlreiche
weitere Beobachtungen erhärtet und stellt uns in Gegensatz zu
Vorländer, der zu seiner Theorie über „Harze und Lacke" die Ein¬
trittsstelle in para am Phenylrest forderte, gestützt auf Analysen
der Abbauprodukte des Mononitro-triphenylbenzols. Durch An¬
wendung von geeigneten Trennungsmethoden waren wir imstande,
neun verschiedene Nitrierungsprodukte des Triphenylbenzols zu
isolieren, worüber Abschnitt 2 berichtet.
1. Nitrierung von Tr i p h e n y 1 b e n z o 1
Triphenylbenzol ist in Schwefelsäure unlöslich. Es ist deshalb
nicht möglich, in Schwefelsäure nach den bekannten, zur Kontrolle
der Nitrierung besonders geeigneten Methoden zu nitrieren. Durch
geeignete Abstimmung von Lösungsmittel, Salpetersäure, Tempe¬
ratur und Reaktionsdauer kann die Nitrierung trotzdem so gelenkt
werden, daß entweder zur Hauptsache (75 o/o) Mononitro-triphe-
nylbenzol oder im extremen Falle nur höher nitrierte Produkte ent¬
stehen. Um die von Meilin und Engler bezeichneten Nitrierungs¬
produkte (zwei Tetranitro-Verbindungen und eine Trinitro-Verbin-
dung) des näheren zu untersuchen, wurden entsprechende Nitrier¬
methoden gewählt.Bei der ersten Methode wurde das Triphenylbenzol nach An¬
gaben von Engler in rauchender Salpetersäure gelöst und 10 Mi¬
nuten in der Wärme behandelt. Durch Eingießen in Wasser fiel

— 26 -
ein gelbliches Produkt in Flocken aus, das sich jedoch als ein
Gemisch erwies. Durch Extraktion mit Alkohol blieb ein Rückstand
von 18 o/o vom Schmelzpunkt 151 — 193°, der nicht weiter unter¬
sucht wurde. Die alkohollöslichen Nitrierungsprodukte wurden
wie im nächsten Abschnitt angegeben aufgearbeitet.Bei der zweiten Nitriermethode wurde das Triphenylbenzol in
heißem Eisessig gelöst und mit einem Überschuß mit 70proz. Sal¬
petersäure während 2 Stunden nitriert, um die von Meilin ange¬
gebenen zwei Tetranitro-Verbindungen zu reproduzieren. Tatsäch¬
lich war das oc-Tetranitrotriphenylbenzol im in Eisessig unlöslichen
Teil eindeutig zu isolieren, neben einer zweiten, bisher unbekann¬
ten, schwer löslichen isomeren Tetranitro-Verbindung, während
sich die von Melliti bezeichnete isomere ^-Verbindung im löslichen
Teil als ein Gemisch verschiedener Produkte erwies, die durch
ausgedehnte fraktionierte Kristallisationsversuche nicht zu trennen
waren. Die aufgefundene, in Eisessig lösliche, aber in Alkohol un¬
lösliche Tetranitrotriphenylbenzol-Verbindung II ist in kleinen
Nadeln aus Chlorbenzol erhältlich. Es handelt sich dabei um eine
zu oc-Tetranitrotriphenylbenzol isomere Meta-Verbindung.Schließlich wurde eine Nitrierung in 75proz. Salpetersäure
angesetzt, um auf diese Weise die gewünschten Trinitro-Verbin-
dungen zu erhalten. Bei dieser Nitrierung war aus dem in Alkohol
unlöslichen Teil ebenfalls das oc-Tetranitrotriphenylbenzol neben
der oben erwähnten in Alkohol unlöslichen isomeren Meta-Verbin¬
dung zu isolieren, während aus dem in Alkohol löslichen Teil der
Nitrierungsprodukte zwei weitere isomere Tetranitro-Verbindun¬
gen zu erkennen waren.
2. Trennung der Nitrierungsgemische nach der
chromatographischen Adsorptionsmethode
Die Vielzahl der möglichen isomeren Verbindungen der Tri-
phenylbenzol-Nitrierung, wie auch die schlechte Kristallisations¬
fähigkeit der in Alkohol löslichen Produkte erschwerten die Iso¬
lierung einzelner Glieder aus den Gemischen. Sorgfältige Versuche,mit fraktionierter Kristallisation zum Ziele zu gelangen, scheiter-

— 27 —
ten an den schlechten Kristallisationsfähigkeiten der löslichen Pro¬
dukte. Wir wählten schließlich nach einigen Erfolg versprechendenVorversuchen die chromatographische Adsorptionsmethode zur
Trennung der Nitro-Gemische, obwohl die Anwendung dieser prä-
parativen Methode zur Aufarbeitung von Nitrierungs-Gemischenin der Literatur nicht aufzufinden war. Die Anforderungen, die im
vorliegenden Falle an diese Methode gestellt werden, nämlich die
Prüfung eines Gemisches auf Einheitlichkeit und die Isolierung der
Komponenten, liegen aber gerade im Anwendungsbereich der chro¬
matographischen Adsorptionsmethode, die auf dem Gebiete der
chemischen Gemische, wie sie bei Synthesen und Abbauverfahren
entstehen, und auch in Naturprodukten vorliegen, heute nicht weg¬
zudenken ist. Nach den bisherigen Erfahrungen begünstigen unge¬
sättigter Charakter, polare Gruppen und Molekülgröße des Stoffes
die Adsorption an hydrophilen Adsorptionsmitteln. Bei Nitrotri-
phenylbenzolen sind diese Voraussetzungen in günstigem Maße
erfüllt, so daß ein Gelingen dieser Methode anzunehmen war.
Arnold [40] stellt in seinen Versuchen an Nitrophenol und Nitra-
nilin, fest, daß bei isomeren Molekülen mit gleichen funktionellen
Gruppen die isomere Verbindung mit größerem Dipolmomentestärker adsorbiert wird. In den untersuchten Produkten nimmt
die Stärke der Adsorption von ortho nach para zu.
Zur Anwendung gelangte das Durchlauf-Chromatographier-verfahren. Als Adsorbens diente stets eine Aluminiumoxydsäule
vom der Aktivität 1—2 [41]. Entwicklet wurde mit Petroläther oder
Petroläther-Benzol-Gemischen, um dann mit Lösungsmitteln stär¬
kerer Affinität zu Aluminiumoxyd, wie sie aus der experimentellen
Erfahrung bekannt sind, die Stoffe zu eluieren [42].In der vorliegenden Arbeit wurde die chromatographische
Trennungsmethode so weit zur Anwendung gebracht, daß die Viel¬
gestaltigkeit der entstandenen Nitrierungsprodukte eindeutig er¬
wiesen werden konnte. Es gelang, aus den alkohollöslichen Ge¬
mischen mindestens sieben Komponenten zu isolieren, während
bisher nur zwei verschiedene Stoffe — wovon einzig das Mono-
nitrotriphenylbenzol eindeutig bezeichnet — zu fassen waren. Die
Möglichkeiten, weitere Nitroverbindungen aus den Gemischen zu
isolieren, sind damit noch keineswegs erschöpft.

— 28 —
a) Trennung der löslichen Bestandteile der 1. Nitrierung
Es wurde über eine Aluminiumoxyd-Säule vom 20fachen Ge¬
wicht der Substanz mit Aktivität 1—2 chromatographiert. Die
Substanz war in Petroläther zu wenig löslich, so daß mit Petrol-
äther-Benzol (1:10) entwickelt und mit Benzol eluiert werden
mußte. Im entwickelten Chromatogramm waren, vor allem drei.
Zonen zu erkennen, was das Vorhandensein eines Gemisches deut¬
lich erwies. Bei den Fraktionen 3/6/10 verließen je eine deutlich
sichtbare Zone die Säule. Die Hauptmenge des Gemisches befand
sich in der zweiten Zone, während in der ersten Zone 21 o/0 und in
der dritten Zone 17 o/o vorhanden waren. Zur Untersuchung ge¬
langten jedoch nur die Produkte der ersten und zweiten Zone. Die
Analysenresultate der eluierten Substanzen deuten auf höher
nitrierte Produkte mit 5—6 Nitrogruppen im Molekül hin. Mit
einem erneuten Chromatogramm 5) der Fraktionen war eine ein¬
heitliche Substanz als Pentanitrotriphenylbenzol zu isolieren, wäh¬
rend für eine weitere Fraktion die Hexanitro-Verbindung zustim¬
men dürfte.
b) Trennung der löslichen Bestandteile der 2. Nitrierung
Das alkohollösliche Gemisch aus der Nitrierung in Eisessigist in Petroläther-Benzol (3:1) löslich und wurde damit auf der
Aluminiumoxyd-Säule entwickelt. Auch in diesem Chromatogramniwar das Wandern von vier deutlich verschiedenen Zonen sichtbar.
(Die den Zonen entsprechenden Fraktionen wurden jeweils zusam¬
mengenommen.) In dem vorliegenden Nitriergemisch überwiegendie niederen Nitrierungsstufen (Mono- und Dinitro-Verbindungen)im Gegensatz zum vorangehenden Chromatogramm. Über Er¬
warten groß war der Anteil desMononitro-Produktes mit 40o,0.
Es sei festgehalten, daß in keinem Chromatogramm eine isomere
Mononitro-Verbindung aufgefunden werden konnte, was die An¬
nahme des einheitlichen Eintrittes der Nitrogruppe unterstützt. —
5) Die Nitrierungsgemische waren nach dem ersten Chromatogrammin keinem Falle in einheitliche Produkte getrennt. Es wurden daher bei allen
Chromatogrammen von den zusammengelegten Fraktionen weitere Chro-
matogramme durchgeführt.

- 29 -
Ebenfalls über einen Drittel war der Anteil der Dinitro-Produkte,
während die höher nitrierten Trinitro- und Tetranitro-Verbindun-
gen in geringeren Mengen von 3 bzw. 8 o/o auftraten. Die Analysen¬werte der vereinigten Fraktionen der zusammengehörigen Pro¬
dukte erweisen wohl die Zahl der vorhandenen Nitrogruppen, doch
zeigen sie noch keine einheitlichen Produkte an. Aus diesem
Grunde wurden auch hier mit den gesammelten Fraktionen weitere
Chromatogramme aufgestellt, um die Stoffe noch besser zu tren¬
nen. Die durch erneutes Chromatographieren an Aluminiumoxyd
erhaltenen Produkte zeichnen sich mit Ausnahme der Mononitro-
Verbindung durch schlechte Kristallisationsfähigkeit aus.
Die Tatsache, daß Nitro-Verbindungen überhaupt nicht zur
Kristallisation gebracht werden können, sondern stets in lack- oder
glasartigen, aber doch ähnlich struierten Teilchen aus den Lösungs¬
mitteln erhalten werden, beobachteten Vorländer und Mitarbeiter
an verschiedenen Verbindungen [43]. So erwähnt der Verfasser in
einem Bericht („Die Überkühlung amorpher Schmelzen im Zu¬
sammenhange mit der molekularen Gestalt") monomolekulare
Substanzen, die in kristallin festem Zustande überhaupt nicht exi¬
stieren, sondern nur in Form von harz- oder gummiähnlichen,unterkühlten Schmelzen. Als auffälligste Beispiele werden Tri-
(nitrophenyl) -carbinol bzw. Tri-(nitrophenyl)-methylcyanid er¬
wähnt, Substanzen, die in struktureller Hinsicht mit den Nitrotri-
phenylbenzolen weitgehende Ähnlichkeit aufweisen. Vorländer
schreibt die Unfähigkeit der Moleküle sich im Raumgitter anzu¬
ordnen, d. h. zu kristallisieren, in erster Linie der verzweigten und
dissymmetrischen Gestalt des Moleküls zu. Diese Substanzen
weisen überdies auch keinen scharfen Schmelzpunkt auf, sondern
erweichen zunächst, werden durchsichtig und zerfließen schlie߬
lich, um dann beim Erkalten eine überkühlte Schmelze zu bilden,
die nicht zur Kristallisation gebracht werden kann.
Die eluierten Substanzen (ausgenommen das Mononitrotri-
phenylbenzol) konnten in keiner Weise zur Kristallisation gebracht
werden, sondern erstarrten aus den Lösungsmitteln stets in glas¬
artigen, ähnlichen Teilchen, die keinen scharfen Schmelzpunkt auf¬
wiesen, sondern einen oben beschriebenen Schmelzvorgang durch¬
machten.

— 30 —
c) Trennung der löslichen Bestandteile der 3. Nitrierung
Das Chromatogramm der in Alkohol löslichen Produkte weist
auf die Anwesenheit von hauptsächlich zwei Tetranitro-Verbin¬
dungen hin. Die beiden Verbindungen konnten in Benzol-Alkohol
ebenfalls nicht zur Kristallisation gebracht werden, sondern wur¬
den in glasartigen, doch ähnlich struierten Teilchen erhalten, was
nach Vorländer [43] auf verzweigten, dissymmetrischen Bau
schließen läßt. Wie es für solche Substanzen zu erwarten ist, warenkeine scharfen Schmelzpunkte vorhanden. Beim Erwärmen er¬
weichten die beiden Tetranitro-Verbindungen, wurden durchsichtigund schließlich flüssig. Die Analysenresultate waren mit den be¬
rechneten Stickstoffwerten in Übereinstimmung.
Nitrierung von Triphenylbenzol
N itrierung in
Nitrierungsprodukt
Eisessig +HN03(1,52)
HN03(1,42) HN03(1,52)
Alkohol Alkohol Alkohollösl. 1 unlösl. lösl.
%unlösl. lösl. i unlösl.
<7o 1 °/o
Mononitro -Triphenylbenzol
1
40*)Dinitro -Triphenylbenzol 36
Trinitro -Triphenylbenzol 3
a-Tetranitro -Triphenylbenzol 2 16
Tetranitro-Triphenylbenzol II 1,5 33**)Tetranitro-Triphenylbenzol III 8
Tetranitro-Triphenylbenzol IV 30
Tetranitro-Triphenylbenzol V 12
Pentanitro -Triphenylbenzol 21
Hexanitro -Triphenylbenzol 32
Nicht untersuchte Produkte
und Verluste 9,5 29 18 9
*) Die Prozentangaben verstehen sich als Gewichtsprozente und be¬
ziehen sich auf die rohen Nitrierungs-Gemische.**) Die Nitrogruppe tritt mehrheitlich in meta-Stellung der Phenyl-
reste ein, während etwa bei der Nitrierung von Diphenyl in meta praktischkeine Substitution erfolgt.

— 31 —
3. Zusammenstellung der Nitrierungs-Ergebnisse
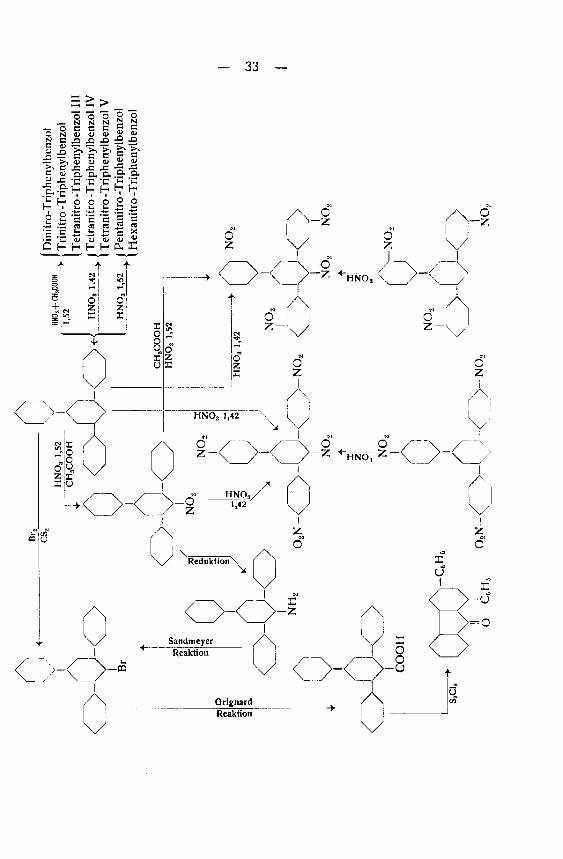
Durch die Nitrierung von Triphenylbenzol konnten zehn ver¬
schiedene Nitrierungs-Prödukte erhalten werden. Von diesen 10
Verbindungen waren einzig 4 in kristallinen Substanzen erhältlich,
während die 6 übrigen nicht zur Kristallisation gebracht werden
konnten. Die ungenügenden kristallinen Eigenschaften der Nitro¬
verbindungen erschwerten eine eindeutige Identifizierung außer¬
ordentlich.
Aus der Tabelle ist ersichtlich, daß unter den gewählten
Nitrierungs-Bedingungen keine Produkte zu mehr als 40o/0 erhält¬
lich sind, d. h. bei der Nitrierung ist keine Dirigierung an be¬
stimmte, ausgezeichnete Stellen des Moleküls vorhanden, wenn
vom Eintritt der 1. Nitro-Qruppe in den zentralen Kern abgesehenwird.
4. Konstitutionsermittlung einiger Nitrierungs-
produkte des Tripheny 1benzo1 s
Über die bisher bekannten Nitrierungsprodukte des Triphenyl-benzols liegt einzig eine Konstitutionsermittlung des Mononitro-
Derivates von Vorländer [43] vor, sowie eine kurze Angabe vom
gleichen Verfasser über das ot-Tetranitrotriphenylbenzol. Im fol¬
genden sei ein weiterer Beitrag zur Konstitutionsaufklärung dieser
Produkte erwähnt.
a) Mononitrotriphenylbenzol
Vorländer versuchte die Konstitutionsermittlung des Mono-
nitrotriphenylbenzols durch oxydativen Abbau aufzuklären. Bei
Oxydationsversuchen in Eisessig mit Chromsäure erhielten Vor¬
länder und Mitarbeiter 12 o/o Benzoesäure, 4 o/0 p-Nitrobenzoesäureund etwa 20 o/o einer zweibasischen Säure, welche sie als 5-(p-
Nitrophenyl)-benzol- 1,3-dicarbonsäure (I) bezeichneten. Für eine
in Wasser schwerlösliche Substanz, die 56 <y0 des Oxydationspro¬duktes darstellt, gaben sie die Formel 5-(p-Nitrophenyl)-3-phenyl-benzoesäure-1 an. Unsere Beobachtungen und Ergebnisse stehen
aber in Gegensatz zu den obigen Autoren, und da dieselben eine

— 32 —
Teilsynthese ihrer Oxydationsprodukte unterließen, dürfen wir —
ohne die Analysenresultate zu bezweifeln — annehmen, daß die
Nitrogruppe in diesen Verbindungen an anderer Stelle sitzt. Wird
die Nitrogruppe, wie von uns verlangt, im zentralen Kern ange¬
nommen, so sind Oxydationsprodukte mit genau gleichen Summen¬
formeln denkbar. Nur wäre Oxydationsprodukt I als l-Phenyl-2-nitro-3,5-iso-phthalsäure zu bezeichnen und die zweite Verbindungals l,3-Diphenyl-2-nitro-benzoesäure-5. Die Beweiskraft der Kon-
stitutionsermittlung von Vorländer wird dadurch in Frage gestellt.Kohler [44] bewies in seinen umfangreichen Untersuchungen,
daß beim bekannten Monobromtriphenylbenzol das Brom am zen¬
tralen Kern sitzt. Es gelang ihm, nach der Methode von Qrignarddas schwer zugängliche Magnesium-Derivat herzustellen und durch
Einwirkung von Kohlendioxyd Triphenylbenzoesäure herzustellen.
Alle Versuche, aus der Karbonsäure ein Säurechlorid herzustellen,endeten stets mit einer Kondensation zu 1,3-Diphenylfluorenon.Mit einer Carboxylgruppe in para eines Phenylrestes des Tri-
phenylbenzols wäre die Bildung des bekannten Fluorenons aus¬
geschlossen. Überdies beobachtete der Verfasser, daß beim Ersatz
des Broms durch den Acetylrest ein Keton entsteht, das sich wie
ein Methylketon mit zwei Substituenten in o-Stellung verhält. Die
Stichhaltigkeit der Konstitutionsaufklärung des Monobromtriphe-nylbenzols durch Kohler ist augenscheinlich, zumal der Verfasser
Kenntnis von den Arbeiten Vorländers hatte.
Es war für uns nun gegeben, das Mononitro-Derivat in die
Bromverbindung überzuführen und diese mit dem von Kohler be¬
schriebenen (durch direkte Bromierung erhaltenen) Monobromtri¬
phenylbenzol zu vergleichen. Schon das überaus schwach basische
Amin deutete auf eine Stellung im zentralen Kern hin. Es gelangnicht, wie beim Tri-(nitrophenyl)-benzol während der Reduktion in
Eisessig ein N-Acetyl-Derivat zu erhalten, was vor allem auf die
sterischen Verhältnisse zurückzuführen ist. Ebenso deutet die
äußerst schwache Basizität des Amins (Monoaminotriphenylbenzolist in Säuren und Laugen jeder Konzentration unlöslich) in die¬
selbe Richtung. Nachdem wir aber mit dem Ersatz der Amino-
gruppe durch Brom ein in allen Eigenschaften dem von Kohlerbeschriebenen Bromderivat identisches Produkt erhielten, darf an-
- 33 —
III > >
O O O 0 O
N N NN N
_ 0 e C C C 3
c N tu a> <u Q <u
N C .0 X) .Q .O ;03
"?. "?> ?> ">> ">..O c s c C 8
>, aj a> a» <u <u
>> C _c _n X .s J3C <U .2-.-
.-
Q. a. c. C.0)
J3
Cl. riph 'C 'C 'C -Tri -TriTri H 0
1.O 0
u
0 0
01*0 'S 'S 'S s 'E
~*^ ^ es ts « rt «
ni _3 L. w t- ~G X
'1— (U eu aj <u
Q H H H H à E

— 34 —
genommen werden, daß auch bei der Nitrierung zu Mononitrotri-
phenylbenzol die Nitrogruppe im zentralen Kern sitzt.
b) OL-Tetratütrotriphenylbenzol
Bei der Oxydation von x - Tetranitrotriphenylbenzol erhielt
Vorländer stets p - Nitrobenzoesäure und forderte daher für
dieses schwerlösliche Nitro-Derivat eine Konstitution, bei der drei
Nitro-Gruppen in para der Phenylreste stehen und eine im zen¬
tralen Kern. Diese Annahme konnte bestätigt werden, indem wir
sowohl bei der Weiternitrierung des Tri-p-nitrophenyl-benzols wie
auch der Mononitro-Verbindung, die eine Nitro-Qruppe im zen¬
tralen Kern besitzt, identische Tetranitro-Verbindungen erhielten.
Durch diese beiden Nitrierungen wurde die Konstitution des «.-
Tetranitrotriphenylbenzols erhärtet.
c) Tetranitrotriphenylbenzol II
Bei der Aufarbeitung der Nitrierungsprodukte war im alkohol¬
unlöslichen Teil eine Verbindung, die in Eisessig löslich, in Alko¬
hol dagegen unlöslich war und sich als eine Tetranitro-Verbindungherausstellte. Der hohe Schmelzpunkt und die Löslichkeitsverhält-
nisse deuteten auf eine ähnliche Anordnung der Nitrogruppen hin,wie sie bei ^-Tetranitrotriphenylbenzol gefunden wurde. Durch
Weiternitrieren des Tri-m-nitrophenyl-benzols in wenig Eisessigund Salpetersäure (d = 1,52) resultierte ebenfalls ein Tetranitro¬
triphenylbenzol, das sich mit dem aus der Nitrierungsmasse auf¬
gearbeiteten Produkte identisch erwies. Ebenso gelang es, durch
Weiternitrieren der Mononitro-Verbindung dieselbe Tetranitro-
Verbindung zu erhalten, womit auch die Konstitution dieses iso¬
meren Tetranitrotriphenylbenzols II erwiesen sein dürfte.
Zur Konstitution der aus den Chromatogrammen erhaltenen
Nitroprodukte sei erwähnt, daß es durch Weiternitrierung der Di-
und Trinitro-Verbindungen nicht gelang, eines der bekannten
schwerlöslichen Produkte vom Tetranitrotriphenylbenzol-Typus zu
erhalten. Damit scheint es unwahrscheinlich, daß die zweite bzw.
zweite und dritte Nitrogruppe in para oder meta der Phenylrestesitzt.

Experimenteller Teil
I. Darstellung von Triphenylbenzol-Derivatendurch Kondensation von Acetophenonen
1. Ausga n gs p ro du k t e
a) m-Nitroacetophenon
60 g (| Mol) Acetophenon wurden in einem gekühlten Nitrier¬
gefäß langsam in 300 g Schwefelsäure (d = 1,84) in der Weise
gelöst, daß die Temperatur nie über 10° zu stehen kam. In die auf
0° gekühlte Lösung ließ man 90 g Salpetersäure (d == 1,42) ge¬
löst in 100 g konz. Schwefelsäure derart zutropfen, daß die Tem¬
peratur nie über 5° stieg. Das Gemisch wurde noch 2 Stunden
gut gerührt und dann auf 1 kg Eis gegossen, worauf das Nitro-
acetophenon als gelbe Masse ausfiel. Nach dem Umkristallisieren
aus verdünntem Alkohol resultierten 62 g m-Nitroacetophenon
(Smp. = 80°) in zentimeterlangen Prismen, was einer Ausbeute
von 76 o/o entspricht.
b) p-Nitroacetophenon
Bereitung des Natriumacetessigesters : In einen 500 ccm
fassenden Rundkolben mit Rückflußkühler werden 200 ccm über
Natrium getrockneter Äther gebracht. 7,1 g Natrium schüttelt man
in einem Kolben in heißem Xylol, bis sich das Natrium in gleich¬
mäßige, kleine Kügelchen aufgeteilt hat. Nach dem Dekantieren
wäscht man mit Äther und gibt darauf das feinverteilte Natrium
in den vorgelegten Äther. In den eisgekühlten Äther läßt man
40 g frisch destillierten Acetessigester zutropfen, in der Weise,
daß die Reaktion nicht zu heftig wird. Sobald eine weiße Schicht
von Natriumacetessigester um die Natriumkügelchen gebildet ist,
läßt die Reaktion nach und muß durch Erwärmen der ätherischen

— 36 —
Lösung am Rückflußkuhler unterstutzt werden. Nach 12 Stunden
Reaktionsdauer erfüllt eine voluminöse weiße Masse von Natrium-
acetatessigester den Kolben. 58 g gereinigtes p-Nitrobenzoyl-chlorid werden in 120 ccm Äther in der Wärme gelöst und unter
Wasserausschluß dem gekühlten Natriumacetessigester zugegeben.Der voluminöse Natriumacetessigester-Niederschlag lockert sich
sofort, und unter Orange-Färbung beginnt die Reaktion mit p-
Nitrobenzoylchlorid. Dabei fällt Natriumchlorid in kleinen Kri¬
stallen aus, während der p-Nitrobenzoyl-acetessigester sich in
Äther löst. Nach 2stündigem Erhitzen unter Rückfluß wird vom
ausgeschiedenen Natriumchlorid abfiltriert. Im Vakuum wird der
Äther abgezogen, worauf ein hellgelbes, zähflüssiges Öl zurück¬
bleibt, das erst in der Kälte erstarrt.
Erhalten: 77 g p-Nitrobenzoyl-acetessigester.Verseifung des Acetessigesters: Nach Gevekoht [2] läßt sich
p-Nitrobenzoyl-acetessigester mit 33proz. Schwefelsäure oder mit
Alkali verseifen, doch sind die Resultate der Verseifung mit Alkali
unbefriedigend. Der Acetessigester wird in einen 500 ccm fassen¬
den Rundkolben mit Rückflußkühler in 400 ccm 33proz. Schwefel¬
säure gegeben und während 22 Stunden unter Rückfluß erhitzt.Nach dem Erkalten erstarrt das bräunliche Öl zu einer körnigenMasse, welche durch Filtration von der Schwefelsäure getrenntwird. Nachdem die Masse in 200 ccm Äther aufgenommen worden
ist, wird im Scheidetrichter mit Natriumbicarbonat gründlich aus¬
gewaschen, bis keine p-Nitrobenzoesäure mehr nachzuweisen ist.
In der ätherischen Lösung bleibt das p-Nitroacetophenon gelöst.Erhalten: 34 g p-Nitroacetophenon.Durch Vakuumdestillation bei 150° und 9 mm Hg und Um-
kristallisation aus 100 ccm verd. Alkohol sind 25 g reines, hell¬
gelbes p-Nitroacetophenon vom Smp. 81° erhältlich, was einer
Ausbeute von 49 o/0 entspricht.
c) p-Bromacetophenon
p-Bromacetophenon wurde nach der von Groggins [9] be¬
schriebenen Methode aus Brombenzol mit Acetanhydrid in Gegen¬wart von Aluminiumchlorid hergestellt. Die in Äther aufgenom-

- 37 -
mené Reaktionsmasse ergab bei der Destillation das bekannte p-
Bromacetophenon mit einer Ausbeute von 61 o/o. Durch Umkristal-
lisation aus Alkohol erhielt man in Übereinstimmung zu Groggins
eine weiße Substanz in feinen Blättchen vom Smp. 53—54°.
d) m-Bromacetophenon
82 g (| Mol) m-Nitroacetophenon wurden nach der Methode
von Bechamp und Brimmeyer, wie sie in den „Grundlegenden Ope¬
rationen der Farbenchemie" für Chlornitrophenol [45] beschrieben
ist, reduziert. Die Gewinnung des Amins aus der Reaktionsmasse
erfolgte in der Weise, daß das alkalische Gemisch mit dem gleichen
Volumen Alkohol versetzt und heiß vom Eisenschlamm getrennt
wurde. Im Vakuum wurde zur Trockene eingedampft und der Rück¬
stand in Äther aufgenommen. Nach dem Abdampfen des Äthers
waren durch Umkristallisation aus verd. Alkohol gelbe Blättchen
vom Smp. 98° mit einer Ausbeute von 83 o/o erhältlich.
Zum Ersatz der Aminogruppe durch Brom nach der bekannten
Methode von Sandmeyer bereiteten wir eine Kupferbromür-Lösungin folgender Weise:
In einen 750 ccm fassenden Rundkolben mit Rückflußkühler
werden 21 g Kupfersulfat (krist.), 7 g Naturkupfer, 51 g Natrium-
bromid, 10 g Schwefelsäure (1,84) und 10 g 22proz. Bromwasser¬
stofflösung, gelöst in 300 ccm Wasser, gebracht. Das Gemisch
wird nun so lange unter Rückfluß erhitzt, bis die anfänglich blaue
Lösung hellgrün wird und sich gelbliches Kupferbromür ausge¬
schieden hat.
Wenn die Kupferbromür-Lösung bereitet ist, löst man 45 g
(1/3 Mol) Aminoacetophenon in 100 ccm 0,5-n Bromwasserstoff¬
lösung und diazotiert in bekannter Weise bei 0° mit eingestellter
1-n Natriumnitritlösung. Nach 30 Minuten ist die Diazotierung
beendet.
In den die Kupferbromürlösung enthaltenden Rundkolben
wird ein Tropftrichter gesetzt, derart, daß die Trichtermündung
unter die Oberfläche zu stehen kommt. Die Diazonium-Lösung
wird nun während 15 Minuten in die siedende Kupferbromür-
Lösung zugegeben, wobei sich sofort unter heftiger Stickstoffent-

— 38 —
wicklung ein braunes Produkt ausscheidet. Unter Rühren beläßt
man noch weitere 2 Stunden in der Hitze.
Die von der Lösung abfiltrierten Produkte werden in Äther
aufgenommen und mit schwach salzsaurem Wasser ausgeschütteltund gewaschen. Nach erfolgter Vakuumdestillation erhielt man
15 g m-Bromacetophenon als ein gelbliches Öl mit einer Ausbeute
von 23 »o bezogen auf m-Nitroacetophenon. Bei 7° erstarrt das
m-Bromacetophenon zu einer kristallinen Masse.
2. Kondensation von Acetophenon-Derivaten
a) Kondensation mit Kuliumpyrosulfat und Schwefelsäure
Tri-(m-nitrophenyl)-benzol
Von den zahlreichen Kondensationsversuchen nach Odell und
Hines [25] seien diejenigen mit den besten Ergebnissen erwähnt:
27,5 g (V6 Mol) m-Nitroacetophenon werden mit 35 g frisch
geglühtem Kaliumpyrosulfat in der Reibschale zerrieben und in
einen 100-ccm-Erlenmeyer-Kolben gebracht. In das Gemisch wer¬
den 3,9 ccm Schwefelsäure konz. gut eingerührt, bis eine möglichsthomogene Masse vorliegt. Bei 55° gibt man in den Trockenschrankein und erwärmt während 5 Stunden langsam auf 85° unter ge¬
legentlichem Aufrühren der Masse, um ein Zusammenbacken zu
verhindern. Während der Kondensation verfärbt sich das Gemisch
von Hellgelb nach Braun. Nach weiteren 3 Stunden ist die Kon¬
densation soweit fortgeschritten, daß eine Probe in siedendem
Wasser einen körnigen Rückstand hinterläßt. Es wird weitere 7
Stunden bei 85° belassen, worauf man die Masse in der Reibschale
zerkleinert und zum Herauslösen der anorganischen Substanzenin 700 ccm siedendes Wasser gibt. Es wird mehrmals mit Wasser
extrahiert, bis keine anorganischen Bestandteile mehr nachzu¬
weisen sind. Die zurückbleibende gelbbraune Substanz wird vier¬
mal mit 200 ccm siedendem Alkohol extrahiert, worauf die Kon¬
densations-Zwischenprodukte und harzigen Bestandteile in Lösunggehen.
Der pulverige Rückstand ist in den üblichen organischenLösungsmitteln unlöslich. Das 18 g wiegende Rohprodukt wird

- 39 —
daher in 400 ccm o-Dichlorbenzol heiß gelöst und bei 170° fil¬
triert, da beim Abkühlen unter 150° Trinitro-triphenylbenzol sofort
in kleinen Nädelchen auskristallisiert. Die in gelblichen Nadeln
erhaltene Substanz wird erneut in o-Dichlorbenzol gelöst, mit
Aktivkohle behandelt und umkristallisiert. Es resultierten 15,5 g
schwach gelb gefärbtes Tri-(m-nitrophenyl)-benzol mit einer Aus¬
beute von 64o/o vom Smp. 297—298°.
Zur Analyse wird nochmals in o-Dichlorbenzol mit Tierkohle
behandelt und umkristallisiert, was zu einem fast weißen, in mikro¬
skopisch kleinen Nädelchen anfallenden Produkt vom Smp. 301 —
3020 führt.
16,99 mg gaben 40,76 mg C02 und 5,31 mg H20
C24H15N306 ber. C 65,30% H 3,43% N9,52\
gef. C 65,47 % H 3,50 % N 9,64 %
17,77 mg gaben 1,55 ccm N2 bei 24° und 728 mm
Tri-(m-nitrophenyl)-benzol ist in tiefsiedenden organischen
Lösungsmitteln so gut wie unlöslich, wenig löslich in siedendem
Eisessig und zunehmend löslich in heißem Xylol, o-Dichlorbenzol,
Nitrobenzol und ot-Chlornaphthalin.
Tri-(p-nitrophenyl)-benzol
Die Kondensation von p-Nitroacetophenon erfolgte in ana¬
loger Weise wie oben beschrieben:
10 g p-Nitroacetophenon werden mit 14 g Kaliumpyrosulfat
fein pulverisiert und unter Zugabe von 1,5 ccm konz. Schwefel¬
säure gut vermischt. Die Kondensationsdauer ist gegenüber dem
obigen Versuche auf 17 Stunden auszudehnen und die Temperatur
um 8° auf 93° zu erhöhen. Man gibt ebenfalls bei 55° ein und
beläßt bei dieser Temperatur während 4 Stunden und steigert dann
innert 5 Stunden auf 93°. Die Masse verfärbt sich stärker und
nimmt dunkelbraune Farbe an. Es wird, wie bekannt, mit Wasser
von den anorganischen Teilen befreit und mit Alkohol heiß extra¬
hiert, wobei der Hauptteil des Kondensationsgemisches mit Alko¬
hol in Lösung geht. Als Rückstand verbleibt ein braunrötliches,
körniges Rohprodukt, das in allen gebräuchlichen organischen

— 40 —
Lösungsmitteln sehr schwer löslich ist und sich selbst in siedendem
oc-Chlornaphthalin nur wenig löst. Aus dem grünlich fluoreszie¬renden alkoholischen Filtrate fällt beim Abkühlen ein brauner,harziger Niederschlag aus, der weiter unten (S. 41) Gegenstandeingehender Untersuchung bildet.
Das rohe Tri-(p-nitrophenyl)-benzol wird in 200 ccm sc-Chlor-
naphthalin gegeben, bei 250° gelöst und mit Aktivkohle behandelt.Durch mehrfaches Umkristallisieren aus dem Lösungsmittel wird
schließlich mit erheblicher« Verlusten ein gelbliches Produkt miteiner Ausbeute von 24 o/0 in gebogenen kleinen Nädelchen erhalten,dessen Smp. über 350° liegt. Zur Analyse wird nochmals in x-
Chlornaphthalin mit Aktivkohle behandelt und umkristallisiert.
16,95 mg gaben 1,49 ccm N2 bei 24° und 730 mm
C24H1606N8 ber. N9,52%gef. N9,70%
Tri-(p-nitrophenyl)-benzol ist in o-Dichlorbenzol und tiefer¬siedenden Lösungsmitteln sehr schwer löslich und löst sich in
Nitrobenzol und oc-Chlornaphthalin nur sehr wenig.
Tri-(p-bromphenyl)-benzol
9 g p-Bromacetophenon und 15,5 g Kaliumpyrosulfat werdenin bekannter Weise mit 1,3 ccm konz. Schwefelsäure gemischt und
9 Stunden bei 85° gehalten. Man befreit von den anorganischenSubstanzen durch Extraktion mit heißem Wasser und entfernt die
öligen Bestandteile durch Extraktion mit 200 ccm heißem Alkohol.Als Rückstand verbleibt ein rotbraunes, körniges Rohprodukt, dasin Chlorbenzol aufgenommen wird. Nach Behandeln mit Aktiv¬
kohle und Umkristallisieren aus Chlorbenzol wird Tri-(p-brom-phenyl)-benzol in weißen Nadeln vom Smp. 247—248° erhalten.
19,38 mg gaben 37,65 mg C02 und 4,94 mg H20
C24H16Br3 ber. C 53,07% H 2,78%gef. C 53,02% H 2,85%
Die p-Tribrom-Verbindung ist in Aceton, Alkohol und Ätherschwer löslich, mäßig löslich in Essigester und löslich in Chlor¬
benzol, Xylol und Dichlorbenzol.

— 41 —
Tri-(m-bromphenyl)-benzol
8 g m-Bromacetophenon, 17g Kaliumpyrosulfat und 1,1 ccm
konz. Schwefelsäure werden innig gemischt und wie oben erwähnt
8 Stunden bei 85° kondensiert. Die Masse verfärbt sich zunächst
nach grün und nimmt dann dunkelbraune Farbe an. Das Reaktions¬
gemisch ist nach der Kondensation stets von öligen Bestandteilen
begleitet. Man extrahiert mit Alkohol und befreit von den anor¬
ganischen Substanzen durch Auskochen in Wasser. Zurück bleibt
ein körniges, bräunliches Rohprodukt, das in Essigester aufgenom¬
men wird. Nach Behandlung mit Aktivkohle kristallisiert schlie߬
lich die Tri-(m-bromphenyl)-benzol-Verbindung in weißen Blätt¬
chen vom Smp. 172—173° aus.
20,36 mg gaben 39,53 mg C02 und 4,90 mg H20
C2+H15Br3 ber. C 53,07 °/0 H2,78°/0
gef. C 52,99% H 2,69%
Tri-(m-bromphenyl)-benzol ist löslich in Essigester, Benzol,
Chlorbenzol und Xylol, schwer löslich in Alkohol und Äther.
Kondensation von Dypnon mit p-Nitroacetophenon
3,6 g Dypnon, 2,7 g p-Nitroacetophenon, 10 g Kaliumpyro¬
sulfat und 0,8 ccm konz. Schwefelsäure wurden in analoger Weise
nach den für p-Nitroacetophenon gemachten Angaben kondensiert.
Die Reaktionsmasse verfärbte sich rasch von braun nach schwarz¬
braun. Nach dem Extrahieren mit Alkohol und Eisessig blieben
150 mg eines rötlich-braunen Produktes zurück, das sich nach
Umkristallisation aus a-Chlornaphthalin als Tri-(p-nitrophenyl)-benzol erwies. Aus dem Eisessig-Extrakt gelang es durch wieder¬
holtes Behandeln mit Aktivkohle 100 mg einer Substanz vom
Smp. 169° zu isolieren, die sich mit dem bekannten Triphenyl-
benzol identisch erwies.
Aus den übrigen öligen Produkten war das gesuchte Mono-
nitrotriphenylbenzol nicht zu isolieren.

— 42 —
b) Kondensation in Anilin mit Salzsäure
Wie erwähnt, eignet sich diese Methode vorzüglich zur Kon¬
densation von Acetophenonen mit „neutralen" Substituenten, wäh¬
rend sie bei der Kondensation von Nitro- oder Aminoacetophe-nonen unbrauchbare Resultate zeitigte. Nach dieser Methode
wurde deshalb einzig das Triphenylbenzol als Ausgangsmaterialfür die Nitrierungsversuche dargestellt:
60 g (1/2 Mol) Acetophenon werden in 60 g wasserfreiem
Anilin gelöst und in einen 500-ccm-Langhalskolben gebracht. In
die Lösung gibt man 3 g frisch sublimiertes Anilinchlorhydrat undleitet an die Oberfläche der Lösung trockenes Kohlendioxydgasein. Im Ölbad wird bis zum Siedepunkt des Anilins erhitzt. Bei
180° destilliert aus dem Kolben deutlich sichtbar Wasser ab, was
den Beginn der Kondensationsreaktion erkennen läßt. Nach ein¬
stündigem Erhitzen richtet man den Kolben zur Destillation ein,um vom überschüssigen Anilin zu befreien. Nach dem Abdestil-
lieren von 50 g Anilin bleibt im Kolben ein dunkelroter Rückstand,der in der Kälte zähflüssig wird. Die Masse wird in 300 ccm
heißem Eisessig aufgenommen, worauf beim Abkühlen der Lösung21 g stark verunreinigtes Triphenylbenzol in Nadeln ausfällt. Man
filtriert ab und wäscht mit Eisessig. Aus der eingeengten Mutter¬
lauge werden weitere 6 g erhalten, so daß das Triphenylbenzol-Rohprodukt 27 g (54<y0) beträgt. Zur Reinigung wird die Sub¬
stanz in Eisessig mit Aktivkohle behandelt, bis ein schwach gelb¬liches Produkt vom Smp. 171° in schönen Nadeln auskristallisiert.
Der Schmelzpunkt stimmt mit den bekannten Literaturangabenüberein.
c) Nebenprodukte bei der Kondensation von p-Nitroacetophenon
Bei der Selbstkondensation von p-Nitroacetophenon, die nicht
so günstig verläuft wie bei der isomeren m-Verbindung, bilden die
alkohollöslichen Nebenprodukte den Hauptteil der Kondensations¬
masse.
Durch Einengen und Kühlen des Alkoholextraktes fielen die
löslichen Bestandteile als flockiger Niederschlag aus. Der braune,

- 43 —
harzige Niederschlag wurde getrocknet und pulverisiert. In konz.
Schwefelsäure löste sich eine Probe mit schwach grüner Fluo¬
reszenz. Versuche, die Substanzen durch Umkristallisation zu
trennen und zu reinigen, waren nicht vielversprechend. Bessere
Resultate zeigte die Anwendung des Durchlauf-Chromatographier-verfahrens. Es wurden daher 1,3 g getrocknete Substanz in Petrol-
äther-Benzol 3: 1 aufgenommen und über eine mit Petroläther
bereitete Säule aus 20 g Aluminiumoxyd (Aktivität — 1—2) chro-
matographiert. Die Hauptmenge wurde mit Petroläther-Benzol-
Gemischen eluiert.
Chromatogramm
Fraktion Lösungsmittel j eluierte Substanz
1-2
3-5
6-9
10
200 ccm Petroläther-Benzol 2:1
500ccm Petroläther-Benzol 1:2
600 ccm Äther-Benzol 1:1
200 ccm Äther
280 mg Kristalle, Smp. 138-141 »
805 mg Kristalle, Smp. 118-130°
145 mg Harze
60 mg Öl
Zur weiteren Reinigung der Substanz wurden die Fraktionen
1 — 2 nochmals an 10 g Aluminiumoxyd adsorbiert und mit Petrol-
äther-Benzol-Gemischen 2: 1 eluiert. Aus der ersten Fraktion
resultierte eine in gelben Prismen aus Benzol/Alkohol gut kristal¬
lisierende Substanz vom Smp. 155—156°. Ein erneutes Chromato¬
gramm der Fraktionen 3—5 (aus obiger Tabelle) ergab ebenfalls
zu 87 o/o das gut kristallisierende Produkt. Die Analysen stimmen
auf die Formeln von p,p'-Dinitrodypnon.
20,43 mg gaben 46,12 mg C02 und 7,16 mg H,0
18,92 mg gaben 1,53 ccm N2 bei 23° und 729 mm
C16H12OäN2 ber. C 61,54% H 3,87°/» N 8,97 %
gef. C61,61°/„ H3,92°/o N8,95%
Die aus dem Alkoholextrakt ausgefallenen Nebenprodukte der
Kondensation enthalten somit 74 o/0 Dinitrodypnon. Dinitrodypnon
ist löslich in Aceton, Essigester, Benzol, weniger löslich in Äther.
Mit Schwefelsäure entsteht keine Fluoreszenz. — Da die Äther-
und Benzol-Eluate teilweise ölig herauskamen, wurde auf eine ein-

— 44 -
gehendere Untersuchung der Fraktionen 6—10 verzichtet. Ent¬
sprechend dem Kondensationsverlauf dürfte es sich um Polymeri¬sate von Dinitrodypnon handeln.
Semicarbazon von p,p'-Dinitrodypnon
Zur Darstellung des Semicarbazons wurde eine von den
üblichen Methoden [46] etwas abweichende Darstellungsweise an¬
gewandt:150 mg Dinitrodypnon werden in 0,4 ccm Pyridin gelöst und
mit einem kleinen Überschuß von Semicarbazidhydrochlorid, gelöstin wenig Pyridin, versetzt. Man erhitzt 15 Minuten am Wasserbad
auf 70° und verdünnt mit Wasser, um dann aus Pyridin das Semi¬
carbazon zu kristallisieren. Das Semicarbazon wird in kleinen
Blättchen vom Smp. 226—227° (unter Zersetzung) erhalten.
4,95 mg gaben 10,04 mg C03 und 1,78 mg H20
C17H15OäN5 ber. C 55,28% H 4,09%
gef. C 55,35 X H 4,02 °/o
3. Tri-(aminophenyl)-benzol
a) Reduktion von Tri-(nitrophenyl)-benzolen
Die Reduktion der Nitroverbindungen erfolgte nach zwei
Methoden:
Reduktion mit Zink und Eisessig. 5,5 g Tri-(m-nitrophenyl)-benzol werden in 250 ccm Eisessig, dem 5 ccm Wasser
zugegeben worden waren, gelöst unter Zugabe von 21 g 70proz.Zinkstaub. Die in einem Dreihalskolben mit Rührwerk und Rück¬flußkühler befindliche Reduktionslösung wird auf 115° erwärmt
und während 8 Stunden bei dieser Temperatur kräftig gerührt.Beim Erkalten scheidet sich Zinkacetat in weißen Nadeln ab. Durch
Filtrieren wird vom Zink und Zinkacetat befreit und das Filtratauf 80 ccm eingeengt. Es wird vom restlich ausgeschiedenen Zink¬
acetat abfiltriert, worauf durch Verdünnen des Filtrates mit
Wasser auf 250 ccm ein milchiges Produkt ausfällt, das sich beim
gelinden Erwärmen zu einer grauen „plastischen" Masse zusam-

- 45 -
menballt. Alle Anstrengungen, dieses Produkt durch Lösen in
Alkohol oder Eisessig zur Kristallisation zu bringen, scheiterten.
— (Titrationsversuche in Eisessig und Salzsäure mit 1-n Natrium¬
nitrit-Lösung zeigten, daß keine primäre Aminogruppe im Mole¬
kül vorhanden war. Eine Stickstoffanalyse deutete auf die An-
w esenheit von N-acetylierten Gemischen hin.)
5,3 g des Reduktionsproduktes wurden in 120 ccm 20proz.alkoholischer Kalilauge unter Rückfluß während 8 Stunden ver¬
seift. Nach 3 Stunden schied sich das Amin teilweise in braunen,
feinen Nadeln aus. Nach dem Erkalten filtrierte man ab, engte das
Filtrat im Vakuum auf 30 ccm ein und gab die dreifache Menge
Wasser dazu, worauf das restliche Amin in feinen Nadeln ausfiel.
Zur Aufarbeitung wurde in wenig Alkohol und Salzsäure gelöst,HCl-Gas eingeleitet, bis sich das Chlorhydrat ausschied, filtriert
und der Niederschlag in Wasser gelöst. Durch Zugabe von Ammo¬
niak bis zur alkalischen Reaktion fiel das Amin mit fast weißer
Farbe in Flocken aus und konnte gut filtriert werden. Aus Alkohol
umkristallisiert, erhielt man mikroskopisch feine, lange Nädelchen,
die sich auf der Nutsche verfilzten. Zur Reindarstellung wurde in
Essigester durch Aluminiumoxyd filtriert. — Das analysenreineProdukt schmilzt bei 254° *).
Erhalten: 73 <y0 Tri-(m-aminophenyl)-benzol.
16,22 mg gaben 1,71 ccm N2 bei 19° und 727 mm
C24H21N3 ber. N 11,96 °/o
gef. N 11,79%
Das Amin ist ziemlich stark basisch, löst sich in sehr ver¬
dünnter und konzentrierter Salzsäure. Das Amin ist in Alkohol,
Aceton, Eisessig sehr leicht löslich, in Essigester mäßig löslich
und in Benzol, Xylol, Äther und Ligroin schwer löslich.
Die isomere p-Verbindung wird in gleicher Weise dargestellt.
Tri-(p-aminophenyl)-benzol schmilzt bei 256—257°. Das Amin ist
stärker basisch als die Metaverbindung. Die Löslichkeitsverhält-
nisse sind gleich wie bei der isomeren Verbindung.
!) Zu bemerken ist, daß nicht über Aluminiumoxyd gereinigtes Amin
mit einem scharfen Schmelzpunkt bei 184° schmilzt.

— 46 —
Reduktion mit Zinnchlorür und HCl. Die Re¬
duktion der Nitro-Verbindung erfolgte mit einer Zinnchlorür-
Lösung, bestehend aus 180 g Zinnchlorür in 300 ccm Eisessiggelöst und mit HCl-Gas gesättigt.
11 g Tri-(p-nitrophenyl)-benzol wurden in wenig Eisessiggelöst und mit 300 ccm der Reduzierlösung versetzt. Nach ein¬
stündigem Stehen bei Zimmertemperatur wurde unter Rückfluß
erhitzt unter stetem Einleiten von getrocknetem HCl-Gas in die
Lösung. Nach 3stündiger Reaktionsdauer ließ man erkalten, wor¬
auf sich ein helles Zinndoppelsalz ausschied, das filtriert und mit
wenig Eisessig gewaschen wurde. Das Zinndoppelsalz war in
200 ccm Wasser löslich und wurde langsam in 200 ccm heiße,
30proz. Natronlauge eingerührt, worauf sich das Amin sofort in
hellen Flocken ausschied, während das gebildete Zinnhydroxydin überschüssiger Natronlauge in Lösung ging.
Die Aufarbeitung des Amins erfolgte wie oben beschrieben.
Durch Filtration über Aluminiumoxyd mit Essigester und durch
Umkristallisation aus verdünntem Alkohol erhielt man ein reines
Produkt in feinen, mikroskopischen Nädelchen vom Smp. 256 —
257".
Die isomere Meta-Verbindung wurde in gleicher Weise redu¬
ziert und aufgearbeitet. Die Nadeln wiesen einen Smp. von 254—
255° auf.
b) Derivate der Tri-(aminophenyl)-benzole
B e nzy 1 i d e n-Ve r b in dun g. 70mg Tri-(m-aminophenyl)-benzol werden in 20 ccm Alkohol gelöst und mit 200 mg p-Nitro-
benzaldehyd, gelöst in 5 ccm Alkohol, versetzt. Beim Erwärmen
unter Rückfluß fällt nach 30 Minuten ein gelbes Produkt kristallin
aus. Nach einstündiger Reaktionsdauer filtriert man die erkaltete
Lösung und kristallisiert den Rückstand aus Benzol-Chlorbenzol-
Lösung um. Die Benzyliden-Verbindung wird in gelblichen Blätt¬
chen vom Smp. 257° erhalten.
16,33 mg gaben 1,68 ccm N2 bei 24° und 729 mm
C„HMO.N, ber. N 11,20%
gef. N 11,34%

— 47 —
Die Benzyliden-Verbindung ist in Alkohol, Aceton und Äther
schwer löslich und löst sich zunehmend in Benzol, Xylol, Chlor¬
benzol.
Die isomere p-Verbindung zeigt Farbvertiefung nach hellrot
und schmilzt über 330°. Die Verbindung ist in den entsprechenden
Lösungsmitteln schwerer löslich als das m-Benzyliden-Derivat.
17,80 mg gaben 1,76 ccm N2 bei 20° und 723 mm
Q»HM0,Ne ber. N 11,20%
gef. N 10,96%
p-Toluol sulf amid-Verbindung. 600mgTri-(p-amino-phenyl)-benzol werden in 30 ccm Aceton gelöst und mit 1,1 g
reinem p-Toluolsulfochlorid, gelöst in 10 ccm Aceton, versetzt.
In die Lösung gibt man 0,1 ccm Dimethylanilin und erhitzt unter
Rückfluß. Nach 45 Minuten wird die Lösung mit Natronlaugeschwach alkalisch gemacht und unter Schütteln erkalten gelassen.Von ausgeschiedenen Substanzen wird abfiltriert und das Filtrat
mit verdünnter Salzsäure versetzt, bis ein helles Produkt milchigausfällt. Nach schwachem Erwärmen wird der Niederschlag fil¬
trierbar und in 40 ccm schwach alkalischer Alkohollösung auf¬
genommen. Die Lösung wird durch Filtration geklärt; durch Zu¬
gabe von wenig Salzsäure bis zur kongosauren Reaktion fällt das
Sulfamid in weißen Flocken aus. In Alkohol-Aceton-Lösung wird
mit Aktivkohle behandelt und umkristallisiert. Das Sulfamid kri¬
stallisiert in kleinen, fast weißen Nädelchen vom Smp. 289—290°.
18,38 mg gaben 0,86 ccm N2 bei 23° und 727 mm
C46H8906N8S8 ber. N 5,16%
gef. N 5,16%
Das Toluolsulfamid des Tri-(m-aminophenyl)-benzols wurde
in gleicher Weise dargestellt. Smp. der isomeren m-Verbindung:177—179».
19,60 mg gaben 0,91 ccm N2 bei 21» und 728 mm
C45H3906N3S3 ber. N 5,16%
gef. N 5,16%

— 48 —
Benzoyl-Verbindung. 280 mg Tri-(p-aminophenyl)-benzol werden in 25 ccm Aceton gelöst und mit 0,4 ccm Benzoyl-chlorid im Überschuß versetzt. Man erwärmt 45 Minuten am Rück¬
flußkühler und gibt dann 50 ccm 2-n Natronlauge dazu, worauf
ein weißes Produkt in kleinen Nadeln ausfällt. Die gefällte Sub¬
stanz wird in Alkohol-Essigester-Lösung aufgenommen, mit Aktiv¬
kohle behandelt und auskristallisieren gelassen. Die Benzoyl-Ver¬
bindung sintert bei 156° und schmilzt klar bei 166—168°2).
18,01 mg gaben 1,03 ccm N2 bei 19° und 727 mm
C45H3s03N3 ber. N 6,33%
gef. N 6,40%
Die Verbindung ist löslich in Alkohol, Aceton, Essigester und
Benzol.
Die Darstellung der Benzoyl-Verbindung des isomeren Tri-
(m-aminophenyl)-benzol erfolgte in analoger Weise. Smp. 182—
183».
19,40 mg gaben 1,10 ccm N2 bei 20° und 724 mm
C45H3303N3 ber. N 6,33%
gef. N 6,29%
Hexacetyl-Verbindung. Die Acetylierung wird nach
der bekannten Methode ausgeführt, indem man das Amin in Acet-
anhydrid löst und in der Wärme behandelt, wobei die Lösung1 Stunde bei 100° belassen wird. Beim Kühlen kristallisieren feine,weiße Nadeln aus dem Acetanhydrid. Aus Eisessig kristallisiert
das Hexacetyl-Derivat des Tri-(m-aminophenyl)-benzols in weißen
Nadeln vom Smp. 276° (unter Zersetzung). — Die Ursachen der
zu hohen N-Werte der Analysen wurden im allgemeinen Teil be¬
sprochen.
22,11 mg gaben 1,37 ccm N2 bei 17» und 723 mm
C86H3306N3 ber. N 6,97%
gef. N 7,19%
2) Trotz wiederholter Darstellung des Derivates und gründlicherReinigung liegt der Smp. des aus Alkohol-Essigester-Lösung kristallisierten
Produktes tiefer als bei der entsprechenden isomeren m-Verbindung. Die
Substanz schmilzt zudem nicht wie die übrigen Derivate mit einem scharfen
Schmelzpunkt.

— 49 —
In gleicher Weise erhielt man die isomere p-Verbindung, die
ebenfalls in weißen Nadeln vom Smp. 305—306° aus Eisessigkristallisierte.
16,80 mg gaben 1,09 ccm N2 bei 19° und 724 mm
C36H8306N3 ber. N 6,97%
gef. N 7,23%
c) Ersatz der Amitiogruppen
Ersatz der Aminogruppe durch Brom. Die drei¬
fache Diazotierung des Tri-(aminophenyl)-benzols bot über¬
raschenderweise nicht die geringsten Schwierigkeiten. Man konnte
nach den Methoden, wie sie in den „Grundlegenden Operationender Farbenchemie" für Anilin angegeben sind, verfahren.
349 mg getrocknetes p-Amin werden in 25 ccm Bromwasser¬
stoff-Lösung (2-n) gelöst. Man diazotiert mit eingestellter 1-n
Natriumnitrit-Lösung, die durch eine Mikrobürette langsam in die
eisgekühlte Lösung unter Rühren eingegeben wird. Es wird mit
Natriumnitrit-Lösung bis zur plötzlichen Farbreaktion auf Kalium-
Jodstärkepapier zugegeben.
Verbrauch an 1-n Natriumnitrit-Lösung:
berechnet:: 2,98 ccm
verbraucht: 2,99 ccm.
Der glatte Verlauf der Titration zeigte, daß alle drei Amino-
gruppen des Moleküls ohne gegenseitige störende Beeinflussungsofort diazotiert wurden.
Zum Ersatz der Aminogruppen durch Brom wird eine Kupfer-
bromür-Lösung verwendet, wie sie unter I. l.d) (S. 37) beschrie¬
ben ist. Die Diazonium-Lösung wird tropfenweise unter die Ober¬
fläche der Kupferbromür-Lösung eingeleitet, wobei sich sofort
Stickstoff entwickelt und sich gleichzeitig ein braunes Produkt an
der Oberfläche ausscheidet. Es wird .noch eine halbe Stunde weiter
gekocht und dann von den ausgeschiedenen Produkten abfiltriert.
Durch Extraktion des Rückstandes mit Essigester-Benzol-
Lösung und wiederholtes Behandeln mit Aktivkohle erhielt man

— 50 —
schließlich ein Tri-(p-bromphenyl)-benzol in weißen, kleinen
Nadeln vom Smp. 247—248° in sehr kleiner Ausbeute.
13,31 mg gaben 25,82 mg C02 und 3,30 mg H20
C24H16Br3 ber. C 53,07% H 2,78%
gef. C 52,94 % H 2,77 %
In analoger Weise war die isomere m-Bromverbindung erhält¬
lich, wobei die Aufarbeitung der Reaktionsprodukte mit Essigestersich noch schwieriger gestaltete. Smp. 172—173°.
14,01 mg gaben 27,26 mg C02 und 3,64 mg H20.
C24H18Br3 ber. C 53,07% H 2,78%
gef. C 53,10% H 2,91%
Ersatz der Aminogruppe durch die Azido-
Gruppe. 850 mg Tri-(m-aminophenyl)-benzol werden in be¬
kannter Weise in 30 ccm 1-n HCl dreifach diazotiert, bis zur
Reaktion auf Kalium-Jodstärkepapier. In die bewegte, eisgekühlte
Lösung gibt man tropfenweise 0,75 g Natriumazid, gelöst in
3,3 ccm Wasser, im Überschuß dazu. Unter lebhafter Stickstoffent¬
wicklung scheidet sich ein fast farbloser, körniger Niederschlagan der Oberfläche aus, während gleichzeitig der charakteristische
stechende Geruch der Wasserstoff-Stickstoffsäure wahrnehmbar
ist. Nach 5 Minuten kann bereits keine Diazo-Gruppe mehr nach¬
gewiesen werden. Nach 15 Minuten wird der Niederschlag auf
der Nutsche abgepreßt, mit Wasser und verdünntem Alkohol ge¬
waschen und dann im Vakuum unter Ausschluß von Belichtung
getrocknet. Das Rohprodukt wird mit fast quantitativer Ausbeute
erhalten.
Bereits nach einmaligem Umkristallisieren aus Essigesterwird die Azido-Verbindung in hellen Kristallnadeln erhalten. Eine
zweite Kristallisation aus demselben Lösungsmittel ergibt ein ana¬
lysenreines Produkt vom Smp. 153—154°. Das Azid bleibt im
evakuierten Schmelzröhrchen bis 145° unverändert und nimmt von
dieser Temperatur an dunklere Farbe an, bis zum klaren Schmelz¬
punkt. Oberhalb desselben tritt Zersetzung ein.
10,50 mg gaben 2,78 ccm N2 bei 22° und 729 mm
Q>*H16N9 ber. N 29,36%
gef. N 29,39%

— 51 —
Das Azid der isomeren p-Verbindung kann auf dieselbe ein¬
fache Darstellungsweise erhalten werden. Aus Essigester kristalli¬
siert ein wenig dunkleres Produkt in Nadeln (bis zu 3 mm) vom
Smp. 179—180°.
9,40 mg gaben 2,46 ccm N2 bei 18° und 726 mm
C24HläN9 ber. N 29,36%
gef. N 29,33%
Die Triazide sind leicht löslich in Aceton und Essigester.Unter Belichtung färben sich die Verbindungen langsam von hell¬
gelb nach braun, doch sind sie unbelichtet sehr gut haltbar. (Nach11 Monaten waren keine sichtbaren Veränderungen in Zusammen¬
setzung und Eigenschaften zu bemerken.) In konz. Schwefelsäure
wird sofort Stickstoff in Freiheit gesetzt, während keine Verfär¬
bung auftritt. Durch indirektes Erhitzen auf freier Flamme ver¬
puffen die Azide. Die Azide haben sich zur Charakterisierung der
Amine und zur Analyse durch die leichte Zugänglichkeit und gute
Kristallisationsfähigkeit gut bewährt.
II. Nitrierungsprodukte des Triphenylbenzols
1. Nitrierung des Triphenylbenzols
a) Nitrierung mit rauchender Salpetersäure
Zur Reproduktion der von Engler angegebenen Trinitro-Ver-
bindung wurde die Nitrierung nach folgender Methode durch¬
geführt :
3 g reines Triphenylbenzol vom Smp. 171° werden bei 20°
}n ein Becherglas in 50 ccm HN03 (d = 1,52) eingetragen. Das
Gemisch erwärmt sich unter teilweiser Lösung der Substanz. Man
erwärmt 10 Minuten auf 100°, wobei alles Triphenylbenzol in
Lösung geht und läßt dann erkalten. Die Lösung wird in 250 ccm
Wasser gegeben, worauf das Nitrierungsprodukt in gelben Flocken
ausfällt. Man filtriert, wäscht auf der Nutsche mit Wasser, trocknet
und erhält 4,9 g des rohen Nitrierungsproduktes. Die Substanz
wird 1 Va Stunden mit 400 ccm Alkohol ausgekocht. Nach dem

— 52 —
Dekantieren der gelben alkoholischen Lösung wird noch zweimal
mit Alkohol ausgekocht. Es bleibt ein schwer löslicher Rückstand
von 800 mg zurück (Smp. 151 — 193°), der nicht mehr weiter
untersucht wurde.
Die vereinigten alkoholischen Extrakte werden auf 50 ccm
eingeengt, worauf beim Erkalten gelbe Flocken ausfallen. Die
Mutterlaugen werden wiederholt eingeengt und die Niederschlägevereinigt und getrocknet. Erhalten : 3,8 g alkohollösliche Produkte
vom Smp. 155—250°. Das Nitrierungsgemisch ist in Petroläther
nur teilweise löslich und wird daher zur chromatographischenTrennung in Benzol aufgenommen.
b) Nitrierung in Eisessig-Lösung mit HNOs (1,52)
Zur Darstellung der bisher bekannten oc-Tetranitro-Verbin-
dung forderte Meilin eine Nitrierung in Eisessig, die in folgenderWeise angesetzt wurde :
20 g Triphenylbenzol werden in 200 ccm Eisessig in einem
500 ccm fassenden Dreihalskolben bei 115° gelöst. In die siedende
Lösung läßt man 100 g HN03 (1,52), gelöst in 30 ccm Eisessig,während 15 Minuten zutropfen. (Wie aus einer Probe ersichtlich
war, bildete nach 15 Minuten Monotriphenylbenzol den Haupt¬bestandteil des Nitrierungsgemisches.) Um zu höher nitrierten
Produkten zu gelangen, wird im Ölbad unter Rühren noch U/aStunden bei 120° weiter nitriert.
Während des Abkühlens kristallisiert ein Produkt in feinen
Nadeln aus. Durch Filtration werden 300 mg einer Verbindungerhalten, die über 320° schmilzt. Die Nadeln sind in tiefsiedenden
Lösungsmitteln unlöslich und lösen sich erst in heißem o-Dichlor¬
benzol, Nitrobenzol und a-Chlornaphthalin. Diese Merkmale
deuten darauf hin, daß es sich um das von Meilin beschriebene,schwer lösliche a-Tetranitro-Produkt handelt. Durch Auskochen
mit Eisessig und Umkristallisieren aus o-Dichlorbenzol resultiert
das bekannte a-Tetranitrotriphenylbenzol in schönen, gelbenNadeln.
17,89 mg gaben 1,80 ccm N2 bei 20° und 725 mm
C2iHuO,N4 ber. N 11,52%
•gef. N 11,34%

— 53 -
Zur Fällung der übrigen Nitrierungsprodukte wird die Nitrie-
rungs-Lösung in 350 ccm Wasser gegeben, worauf sich ein flockiger
Niederschlag ausscheidet. Der Niederschlag wird mit Wasser ge¬
waschen, getrocknet und mit 800 ccm Alkohol heiß extrahiert. Die
zurückbleibenden unlöslichen Substanzen werden nochmals mit
250 ccm Alkohol ausgekocht und die alkoholischen Extrakte ver¬
einigt. Der alkoholunlösliche Rückstand beträgt 200 mg. Smp.
255—275°. Nach mehrmaligem Umkristallisieren aus Chlorbenzol
wird ein helles Produkt in feinen Nadeln erhalten, das bei 267—
269° schmilzt: Tetranitrotriphenylbenzol II.
10,58 mg gaben 1,05 ccm N2 bei 19° und 727 mm
CoiHuOsNi ber. N 11,52%
gef. N 11,49%
Die alkoholischen Extrakte werden auf 80 ccm eingeengt.
Beim Erkalten fällt ein flockiger Niederschlag aus, der filtriert,
gewaschen und im Vakuum getrocknet wird. Smp. 75—215°.
c) Nitrieruitg mit HN03 (d = 1,42)
5 g reines Triphenylbenzol werden in 70 ccm 70proz. HNOs
in ein Becherglas eingetragen. Unter Rühren erwärmt man auf dem
Wasserbad auf 85° und beläßt bei dieser Temperatur 20 Minuten.
Nach 4 Minuten geht alles Triphenylbenzol in Lösung, während
sich nach kurzer Zeit gelbe Flocken aus der erwärmten Lösung aus¬
zuscheiden beginnen. Nach dem Erkalten wird in 300 ccm Wasser
gegeben, wobei die Nitrierungsgemische in gelben Flocken aus¬
fallen. Der Niederschlag wird filtriert, mit Wasser gewaschen und
getrocknet. Das 7,0 g betragende Rohgemisch wird in bekannter
Weise mit Alkohol extrahiert.
Als unlösliche Produkte bleiben 3,5 g vom Smp. 255—320°
zurück, welche, wie oben erwähnt, mit Eisessig behandelt werden.
Ein Teil geht in Lösung, während ein Rückstand von 1,1 g ver¬
bleibt, der sich nach dem Umkristallisieren aus o-Dichlorbenzol
mit dem bekannten a-Tetranitrotriphenylbenzol identisch erweist.
Die essigsaure Lösung wird im Vakuum zur Trockne einge¬
dampft, wobei 2,3 g Nitrierungs-Produkte vom Smp. 260—283°

- 54 —
zurückbleiben. Man kristallisiert aus Eisessig und Chlorbenzol um
und erhält ein einheitliches Produkt in feinen Nadeln vom Smp.269°, das mit dem beschriebenen in Eisessig löslichen, in Alkohol
unlöslichen Tetranitrotriphenylbenzol II identisch ist.
Die alkoholischen Auszüge werden, wie vorgängig erwähnt,behandelt und zur chromatographischen Trennung vorbereitet.
2. Trennung der alkohollöslichen Nitrierungs-gemisch e
Die im vorangehenden Abschnitt beschriebenen alkohollös¬
lichen Nitrierungsprodukte stellen stets Gemische verschiedener
Nitro-Verbindungen dar, welche zur Trennung chromatographiertwurden.
Die verschiedenen, je einer sichtbaren Zone zugehörigen Frak¬
tionen der Hauptchromatogramme wurden jeweils vereinigt. Zur
Orientierung über den Grad der Nitrierung wurden von allen ver¬
einigten Hauptfraktionen Analysen gemacht, die im einzelnen nicht
aufgeführt sind, da sie nur als Anhaltspunkte für den Grad der
Nitrierung dienten. Aus den zusammengelegten Fraktionen der
Hauptchromatogramme wurden in keinem Falle analysenreineSubstanzen erhalten. Daher wurde jede Gesamtfraktion einem er¬
neuten Chromatogramm unterworfen, was meistens zu analysen¬reinen Produkten führte.
Mit Ausnahme der Mono- und Trinitro-Verbindungen der
alkohollöslichen Nitrierungsprodukte waren die eluierten Substan¬
zen unter keinen Umständen zur Kristallisation zu bringen, was,
wie bereits früher erwähnt, bei Verbindungen mit verzweigtem,dissymmetrischem Bau oft der Fall ist. — Alle diese nicht kristalli¬
sierenden Substanzen weisen daher auch keinen scharfen Schmelz¬
punkt auf. (Es bleibt die Möglichkeit nicht ausgeschlossen, daß
in den Eluaten der weiteren Chromatogramme zum Teil Gemische
von isomeren Verbindungen vorhanden sein können, die erst durch
weitere, verfeinerte Chromatogramme zu trennen sind. Eine Unter¬
suchung in dieser Richtung ginge jedoch über den Rahmen der
vorliegenden Arbeit.)

— 55 —
a) Trennung der Nitrierungs-Produkte der 1. Nitrierung (S. 51)
1,4 g der alkohollöslichen Nitrierungs-Produkte wurden in
Petroläther-Benzol 1:10 gelöst und an 30 g Aluminiumoxyd (Ak¬
tivität 1—2) adsorbiert.
Chromatogramm 3)
Fraktion Lösungsmittel eluierte Substanz
1—2 100 ccm Benzol 30 mg ölig
3-4 200 ccm Benzol 330 mg nicht kristallisiert
6-9 900 ccm Äther-Benzol 1:1 550 mg nicht kristallisiert
10 400 ccm Äther 270 mg nicht kristallisiert
11—12 300 ccm Äther-Methanol 1:1 20 mg ölig
Fraktionen 3 — 4. Aus Benzol-Äthanol wurden gelbe
Kügelchen vom Smp. 210—280° erhalten. Die Analyse deutet auf
eine Penta- oder Hexanitro-Verbindung hin.
120 mg der Fraktionen wurden erneut an 15 g Aluminiumoxyd
chromatographiert. Mit der ersten Fraktion von 60 mg wurde ein
Produkt erhalten, das aus Benzol-Äthanol in gelben Kügelchen
ausfiel. Smp. 210—222«.
16,35 mg gaben 1,95 ccm N2 bei 24° und 727 mm
C2iH13O10N5 ber. N 13,18%
gef. N 13,11%
Fraktionen 6—9 wurden aus Benzol-Äthanol umkristalli¬
siert. Die Analysenresultate wiesen auf die Anwesenheit von 6
Nitro-Qruppen. Smp. 145—150°.
16,03 mg gaben 2,14 ccm N2 bei 22° und 728 mm
C24H12012N6 ber. N 14,58%
gef. N 14,80 %
s) Die Ergebnisse der Hauptchromatogramme wurden durch die
Arbeiten von Herrn cand. ehem. R. Lieberherr bestätigt.

— 56 —
b) Trennung der Nitrierungs-Produkte der 2. Nitrierung (S. 52)
4 g des alkohollöslichen Nitrierungs-Produktes wurden in
Petroläther-Benzol 3:1 gelöst und auf eine Säule von 80 g Alu¬
miniumoxyd aufgezogen.
Chromatogramm
Fraktion Lösungsmittel eluierte Substanz
2-5
6-9
13
14-15
3600 ccm Petroläther-Benzol 2:1
3500 ccm Petroläther-Benzol 1:1
900 ccm Benzol
300 ccm Benzol
1550 mg kristallisiert
1450 mg nicht kristallisiert
100 mg kristallisiert
300 mg nicht kristallisiert
Fraktionen 2— 5 wurden aus Äther-Äthanol umkristalli¬
siert. Man erhielt gelbe Nadeln vom Smp. 140—142°. Die Ana¬
lyse zeigt die Anwesenheit von einer Nitro-Gruppe im Molekül an.
Der "Schmelzpunkt stimmt annähernd mit dem bekannten Mono-
nitrotriphenylbenzol überein.
150 mg der Mononitro-Verbindung wurden erneut in Petrol-
äther gelöst und an 20 g Aluminiumoxyd adsorbiert. Mit den Frak¬
tionen 2 und 3 des Chromatogrammes wurden 310 mg Substanz
eluiert vom Smp. 142—143«.
18,38 mg gaben 0,66 ccm N2 bei 21° und 726 mm
C24H1702N ber. N 3,99%gef. N 3,98 o/o
Fraktionen 6 — 9 wurden aus Äther-Äthanol umkristalli¬
siert. Der Schmelzpunkt war unscharf von 68—81°. Die Analysedeutete auf die Anwesenheit von 2 Nitro-Gruppen im Molekül.
1001 mg der mit Fraktionen 6—9 eluierten Dinitro-Verbin-
dung wurden in Petroläther-Benzol 4:1 gelöst und an 20 g Alu¬
miniumoxyd adsorbiert. Mit dem gleichen Lösungsmittel im Ver¬
hältnis 3 :1 konnte die Substanz eluiert werden, wobei die Haupt¬menge in der 2. Fraktion erschien. Die Substanz sinterte bei 71°
und wurde bei 83° klar flüssig, während die Analyse die Anwesen¬
heit von 2 Nitro-Gruppen im Molekül bestätigte.

— 57 —
18,37 mg gaben 1,16 can N2 bei 22° und 726 mm
C,4H1604N2 ber. N 7,07%
gef. N 6,98 °/o
Mit Fraktion 13 verließ eine gut erkennbare gelbe Zone
das Chromatogramm. Die eluierte Substanz wies einen wesentlich
höheren Schmelzpunkt auf als die bisher eluierten Produkte und
konnte auch kristallin erhalten werden. Die Analyse sprach für die
Anwesenheit dreier Nitro-Gruppen. Smp. 250—259°. Nach er¬
neuter Adsorption, an Aluminiumoxyd konnte ein Produkt in gelben
Kristallen vom Smp. 263—264° erhalten werden, dessen Analysen¬werte eine Trinitro-Verbindung bestätigten.
17,11 mg gaben 1,44 ccm N2 bei 20° und 729 mm
C24H1506N3 ber. N 9,52%
gef. N 9,41%
Die zusammengelegten Fraktionen 14 und 15 ergaben
aus Benzol-Äthanol ein nicht kristallisierbares Produkt vom Smp.124—175°. Die Analyse deutete auf eine Substanz mit 4 Nitro-
Gruppen hin.
Ein weiteres Chromatogramm der obigen Fraktionen führte
zu besseren Resultaten, wenn auch hier die Substanz nicht kri¬
stallin erhältlich war. Die Substanz sinterte bei 132° und wurde
bei 154° klarflüssig.
14,41 mg gaben 1,42 ccm N2 bei 17» und 726 mm
C24H1408N4 ber. N 11,52%
gef. N 11,48%
c) Trennung der Nitrierungs-Produkte der 3. Nitrierung (S. 53)
2 g des alkohollöslichen Nitrierungs-Gemisches wurden in
Petroläther-Benzol 1:1 gelöst und an 50 g Aluminiumoxyd ent¬
wickelt.Chromatogramm
Fraktion Lösungsmittel eluierte Substanz
1—2 900 ccm Petroläther-Benzol — —
3-5 900 ccm Benzol 1170 mg nicht kristallisiert
6-9 3300 ccm Benzol 450 mg nicht kristallisiert
10-12 650 ccm Benzol-Äther 1:1 200 mg nicht kristallisiert

— 58 —
Fraktionen 3 — 5 wurden aus Benzol-Alkohol umkristalli¬
siert. Die Analysenresultate deuteten auf eine Tetranitro-Verbin-
dung.660 mg des Gemisches wurden erneut an 15 g Aluminiumoxyd
adsorbiert. Die mit Fraktion 4 eluierte Substanz zeigte noch keine
eindeutigen Analysenwerte, die mit Sicherheit auf ein einheitliches
Tetranitrotriphenylbenzol schließen lassen. Immerhin erwiesen
sich die Fraktionen 3—5 des Hauptchromatogrammes von den fol¬
genden Fraktionen durch sichtbare Zonen deutlich verschieden.
Aus Fraktionen 6 — 9 wurde eine Substanz erhalten, die
mit ihren Analysenwerten ebenfalls auf eine Tetranitro-Verbin-
dung hinwies.
560 mg wurden an 15 g Aluminiumoxyd neuerdings chromato-
graphiert, wobei mit Fraktion 4 eine Substanz eluiert werden
konnte, die als eine weitere isomere Tetranitro-Verbindung ange¬
sprochen werden muß. Die Substanz sinterte schwach bei 130°,blieb dann unverändert, um dann langsam von 205—212° klar
flüssig zu werden.
13,9Q mg gaben 1,44 ccm N, bei 23 ° und 726 mm
C24H14N408 ber. N 11,52 %
gef. N 11,34%
3. Ko n s ti t u t io ns e r m i 111 u n g einiger Nitrierungs-Pro dukte
a) Mononitrotriphenylbenzol
Die Nitrierung von Triphenylbenzol zu Mononitrotriphenyl¬benzol erfolgte nach den Angaben von Vorländer [39]:
15 g Triphenylbenzol werden in 145 ccm Eisessig in der Hitze
gelöst und während 15 Minuten bei 118° mit 30 g HN03 (1,52)versetzt. Sofort nach Beendigung des Eintropfens der Salpeter¬säure läßt man unter Rühren erkalten, wobei sich nach 45 Minuten
bei 20° helle, weiße Nadeln auszuscheiden beginnen, welche die
Lösung in einen Brei verwandeln. Man läßt noch 15 Minuten in
der Kälte stehen und filtriert ab.
Es blieben 9,5 g Mononitrotriphenylbenzol zurück, während

- 59 -
durch Einengen der Mutterlaugen weitere 2,5 g gewonnen wurden.
Erhalten: 12 g (= 70<y0).
24,67 mg gaben 0,84 ccm N2 bei 17° und 726 mm
C2iH17OäN ber. N 3,98%
gef. N 3,97%
Reduktion. Die Reduktion erfolgte nach den bereits be¬
schriebenen Methoden mit Zinnchlorür und Salzsäure, sowie mit
Zink und Eisessig. Bei der Reduktion in siedendem Eisessig mit
Zink wurde im Gegensatz zur Reduktion der Tri-(nitrophenyl)-benzole kein Acetylprodukt gebildet.
Das Amin ist sehr schwach basisch und in Mineralsäuren,sowohl ganz schwacher wie starker Konzentration, unlöslich. In
Äther, Alkohol und Eisessig ist das Amin, löslich.
Ersatz der Aminogruppe durch Brom. Infolgeder Schwerlöslichkeit des Amins in Mineralsäuren wurde in Eis¬
essig-Lösung diazotiert.
1 g Monoaminotriphenylbenzol wird in 40 ccm Eisessig in
der Wärme gelöst und mit 4 ccm lOproz. HBr-Lösung versetzt.
Man kühlt rasch auf 20° und gibt die berechnete Menge (3 ccm)1-n Natriumnitrit-Lösung dazu, worauf sich das leichter lösliche
Diazoniumsalz bildet.
Aus 0,7 g Kupfersulfat, 0,2 g Naturkupfer, 1,55 g Natrium-
bromid, 0,2 ccm konz. Schwefelsäure und 20 ccm Wasser wurde
die Kupferbromür-Lösung hergestellt, wie unter 1.1. d) (S. 37)beschrieben.
Die Diazonium-Lösung wurde mit einem Tropftrichter unter
die Oberfläche der siedenden Kupferbromür-Lösung gegeben, wo¬
bei sich sofort Stickstoff entwickelte und eine rötlich-braune Masse
an der Oberfläche ausschied. Es wurde noch eine Stunde unter
Rückfluß weiter gekocht und in der Kälte von den ausgeschiedenenProdukten filtriert. Durch Lösen in 150 ccm Alkohol wurde von
den anorganischen Bestandteilen getrennt. In der Kälte kristalli¬
sierte ein rötliches, in Blättchen auskristallisierendes Produkt aus,
das zur Reinigung dreimal mit Aktivkohle in Äthanol behandelt
und dann in Methanol zur Kristallisation gebracht wurde. Smp.
128,5-1300.

— 60 -
20,50 mg gaben 56,12 mg C02 und 8,16 mg H20
C2iH„Br ber. C 74,81 % H 4,55%
gef. C 74,71% H4,45°/o
Der Schmelzpunkt der Monobrom-Verbindung deutet auf eine
Identität mit dem Monobromtriphenylbenzol, das nach Köhlerdurch Bromieren von Triphenylbenzol in CS2 mit Brom erhalten
wurde: Smp. 129—130°.
19,08 mg gaben 52,29 mg C02 und 7,41 mg H20
C24H17Br ber. C 74,81% H 4,45 °/0
gef. C 74,79% H 4,35%
Die beiden auf verschiedenen Wegen erhaltenen Monobrom-
Verbindungen wurden zur Identifizierung in 10 ccm Alkohol gelöstund zur Kristallisation gebracht. Smp. der gemischten Bromverbin¬
dungen 128,5—129,50.Mit dem Beweis der Identität der beiden Bromverbindungeti
dürfte die hypothetische Annahme des Eintrittes der 1. Nitro-
gruppe in den zentralen Kern des Triphenylbenzols erwiesen sein.
Nitrierung des Mo no ni tr o t r i p h e n y 1 b e n zo 1 s.
0,8 g Mononitrotriphenylbenzol wurden in 6 ccm Eisessig in der
Wärme gelöst. Nach Zugabe von 10ccmHNO3 (1,52) erwärmte man
während 10 Minuten auf 100°. Beim Erkalten fiel das Nitrierungs-gemisch teilweise kristallin aus. Die durch Zugabe von Wasser
gefällten Nitrierungsprodukte wurden dreimal mit Äthanol extra¬
hiert. Als Rückstand blieben 450 mg Substanz. Durch Kochen der
alkoholunlöslichen Produkte in 30 ccm Eisessig ging ein Teil mit
gelber Farbe in Lösung. Das in Eisessig unlösliche Produkt wurde
aus o-Dichlorbenzol umkristallisiert. Smp. über 320°. Das Pro¬
dukt erwies sich mit dem bekannten oc-Tetranitrotriphenylbenzolidentisch, während das eisessiglösliche Produkt mit der isomeren
Meta-Verbindung identisch war. Smp. 267—269°.
b) cL-Tetranltrotriphenylbenzol
Nitrierung von Tri-(p-n itrophenyl)-benzol.410 mg Tri(p-nitrophenyl)-benzol wurden in 4 ccm Eisessig ge¬bracht und langsam mit 5 ccm HNO, (1,52) versetzt. Auf der

— 61
Flamme wurde 5 Minuten zum Sieden erhitzt, bis fast alle Sub¬
stanz in Lösung ging. Beim Erkalten fielen hellgelbe Nadeln aus.
Es wurde in 20 ccm o-Dichlorbenzol gelöst, wobei das unver¬
änderte Ausgangsprodukt unlöslich zurückblieb, während aus dem
Filtrate das bekannte a-Tetranitrotriphenylbenzol mit einem
Schmelzpunkt von über 320° auskristallisierte.
18,51 mg gaben 1,89 ccm N2 bei 18° und 723 mm
C24HU08N4 ber. N 11,52%
gef. N 11,39%
c) Tetranitrotriphenylbenzol 11
In gleicher Weise wurde das Tri-(m-nitrophenyl)-benzolweiter nitriert und ergab nach Umkristallisation aus o-Dichlor¬
benzol ebenfalls ein Tetranitrotriphenylbenzol-Produkt vom Smp.
267—268°, das sich mit dem früher beschriebenen Tetranitrotri-
phenylbenzol II identisch erwies.
17,57 mg gaben 1,8 ccm N2 bei 22° und 737 mm
C24H1408N4 ber. N 11,52%
gef. N 11,50%
d) Reduktion von a-7'etranitrotriphenylbenzol
1 g a-Tetranitrotriphenylbenzol wurde in 70 ccm Zinnchlorür-
Lösung in bekannter Weise reduziert. Das aus dem Zinndoppel¬salz mit Natronlauge 33° Bé gefällte Amin wurde in 300 ccm
Äther aufgenommen. Durch Umkristallisieren aus verdünntem
Alkohol resultierte ein Produkt in feinen Nädelchen, die sich auf
der Nutsche verfilzten. Die mehrmals umkristallisierte Substanz
wies im Gegensatz zu den Angaben von Mellin einen Smp. von
242—244° auf. (a-Tetraaminotriphenylbenzol von Mellin Smp.
138«.)
Sämtliche Mikroanalysen wurden in unserem Mikrolaborato-
rium unter der Leitung von Frl. Dr. E. Pfanner ausgeführt. Ich
möchte der Leiterin dieses Laboratoriums auch an dieser Stelle
für die sorgfältige Ausführung der Analysen bestens danken.

Zusammenfassung
1. Tri-(p-nitrophenyl)-benzol und Tri-(m-nitrophenyl)-benzol, so¬
wie die analogen Bromverbindüngen wurden durch Selbst¬kondensation der entsprechenden Acetophenon-Derivate in
Kaliumpyrosulfat und konz. Schwefelsäure hergestellt.
2. Es gelang, aus den Nebenprodukten der Kondensation von p-
Nitroacetophenon den Hauptbestandteil, das Kondensations¬
zwischenprodukt Dinitrodypnon erstmals zu isolieren und in
das Semicarbazon überzuführen.
3. Zur Charakterisierung der bisher unbeschriebenen Tri-(amino-phenyl)-benzole wurden verschiedene N-Derivate dargestellt.
4. Aus den leicht zugänglichen dreifach diazotierten Verbindun¬
gen konnten eine Reihe von Trisazo-Farbstoffen durch Kup¬peln mit Naphthalin-Derivaten erhalten werden. Die Farbstoffe
weisen ungenügende Affinität zu Baumwolle auf und zeigeneher Eigenschaften entsprechender Monoazo-Farbstoffe.
5. Beim Ersatz der Aminogruppen der Tri-(aminophenyl)-benzoledurch Brom wurden die von uns auf anderem Wege erhaltenen
Tri-(bromphenyl)-benzole erhalten.
6. Durch Versetzen der dreifach diazotierten Verbindungen der
Tri-(aminophenyl)-benzole mit Natriumazid gelang es, die gutkristallisierenden und haltbaren Tri-(azidophenyl)-benzole zu
gewinnen.
7. Durch Nitrierung von Triphenylbenzol konnten unter Anwen¬
dung geeigneter Trennungsmethoden zehn verschiedene Nitro¬
verbindungen isoliert werden, wovon nur vier Verbindungenzur Kristallisation zu bringen waren.

— 63 —
8. Mit dem Ersatz der Aminogruppe des Monoaminotriphenyl-benzols durch Brom wurde eine Monobrom-Verbindung er¬
halten, die sich mit der von, Kohler auf anderem Wege dar¬
gestellten Verbindung identisch erwies.
9. Im Gegensatz zu den bisherigen Anschauungen wurde fest¬
gestellt, daß die erste Nitrogruppe bei der Nitrierung von
Triphenylbenzol in den zentralen Kern des Moleküls eintritt.
10. Die Konstitution von zwei isomeren Tetranitrotriphenylbenzol-
verbindungen konnte sichergestellt werden.

Literaturverzeichnis
1. Etnmerling und Engler, B. 3, 886 (1870); B. 18, 2238 (1885).2. Qevekoht, A. 221, 335 (188..).3. Schweitzer, B. 24, 555 (1891).4. Rupe, B. 34, 3522 (1901).5. Bälow und Hailer, B. 35, 930 (1902).6. Ross, Am. Soc. 59, 1508 (1937).7. — B. 24, 555 (1891).8. Adams, Org. Syntheses V 17.
9. Groggins, C. 36, I, 883 (1936).10. Fierz-David, Grundlegende Operationen der Farbenchemie, 4. Aufl.,
S. 37.
11. Engler, B. 11, 932 (1878).12. Bigelow, Org. Syntheses V, 21.
13. Engler, B. 6, 638 (1873).14. Delacre, Bl. (4) 7, 1041; Ann. chim. (9) 2, 63.
15. Knoll, D. R. P. 250 236.
16. Reddelien, A. 388, 194 (1912).17. Knoevenagel, B. 55, 1929 (1922).18. Porlezza, C. 26, II, 2712 (1926).19. Courtot, Cr. 191, 416 (1930).20. Colonge, Bl. 49, 426 (1931).21. Dolgow, C. 31, II, 3101 (1931).22. Colonge und Grignard, C. 30, II, 1211 (1930).23. Dziewonski, C. 36, II, 1543 (1936).24. Bredereck, B. 72, 1414 (1939).25. Odell und Hines, Am. Soc. 35, 81 (1913).26. Bernhauer und Müller, J. pr. (2) 145, 301 (1936).27. Engler und Dengler, B. 26, 1444 (1893).28. Le Fèvre, Soc. 38, 1467 (1938).29. Reddelien, B. 46, 2716 (1913).30. — Z. angew. Ch. 36, 515 (1923).31. Wuertz, C. 34, I, 2040 (1934).32. Woroshzow, C. 36, II, 2529 (1936).33. - C. 34, II, 1846 (1934).34. Schreiber, Am. Soc. 56, 114 (1934).35. Dübendorfer, Diss. E.T. H. Zürich (1945).

- 65 -
36. Engler, B. 6, 637 (1873).37. Engler und Berthold, B. 7, 1123 (1874).38. Meilin, B. 23, 2533 (1890).39. Vorländer, B. 62, 2836 (1929).40. Arnold, Am. Soc. 61, 1611 (1939).41. Brockmann, B. 74, 73 (1941).42. Trappe, Bioch. Z. 305, 157 (1940).43. Vorländer, Z. physikal. Ch. 105, 246 (1923).
44. Kahler, Am. Soc. 57, 367 (1935).45. Fierz-David, Grundlegende Operationen der Farbenchemie, 4. Aufl.,
S. 67.
46. Staudinger, Anleitung zur qualitativen org. Analyse, 3. Aufl., S. 101.
47. Schultz, Farbstofftabellen, 5. Aufl., S. 21.

Lebenslauf
Am 5. September 1918 wurde ich, Hans Uebersax, Bürgervon Oberönz (Bern) in Zürich geboren.
In Zürich und im LEH Kefikon besuchte ich die Primär- und
Sekundärschulen. Im Frühjahr 1934 trat ich in die technische Ab¬
teilung der Thurgauischen Kantonsschule in Frauenfeld ein und
erhielt daselbst im Herbst 1937 die Maturität. Anschließend stu¬
dierte ich an der Chemischen Abteilung der Eidg. Technischen
Hochschule. Vom Sommer 1939 bis zum Frühjahr 1941 war ich
ununterbrochen zum Militärdienste aufgeboten. Nach weiteren,durch Militärdienst bedingten Unterbrüchen erwarb ich im Herbst
1943 das Diplom als Ingenieur-Chemiker. Mit Anfang des Som¬
mersemesters 1944 begann ich unter der Leitung von Herrn Prof.
Dr. H. E. Fierz-David mit der vorliegenden Promotionsarbeit.
Zürich, den 15. Oktober 1946.





























