Embed Size (px)
Citation preview

Persistence and Persistence and Serendipity!Serendipity!

Requirements for an INDNavigating the Preclinical Sea
FDAGMP
GLP CMC
ICH
IND
COA CFR
Edward Spack, Ph.D.Principal, Vector BioSolutionsManaging Director, Fast Forward LLC
TOX
Clinical TrialResearch

Getting to the Clinic…Without getting lost
“In the middle of my life's journey…. I found myself in a dark wood”
- Dante, Inferno, canto 1
SAMESAFE

Investigational New Drug (IND)When is it required?
• A new chemical entity (NCE) not approved for the indication under investigation
• A drug administered at a new dosage level
• A drug administered with another drug in an unapproved combination
Basic Research
Biology
Target IDTarget
Validation
Chemistry DevelopmentApproved
DrugScreening OptimizationPre-clinical
Phase I
Phase II
Phase III
FDA review
Eureka !
O
OCH3
OCH3
OCH3
OH

Phase 0 Clinical TrialsNot safety, not efficacy- not necessary?
• “Phase Zero” studies are a relatively new approach to initiating clinical trials
• Requires an Exploratory IND (eIND) application
• Minimal requirements (no PK required, single dose tox in rats, CV safety pharmacology in dogs)
• Dose in humans must be non-pharmacologic (i.e., you can’t do efficacy)
• Dose levels very low (typically < 100 µg)
• Useful when there is a particular biomarker to measure, and to obtain blood levels
– Blood levels low, so this may require accelerator mass spectrometry (AMS)
• Preclinical program takes 6-8 months and ~$350K vs. 2 years and $2M+ for IND

Rules for Successful Preclinical DevelopmentBegin with the end in mind
The Clinical End From Target Product Profile (TPP) to NDA to Label Proof of Relevance to Proof of Concept
• competition (current / pending drugs) & market needs• patient sub-populations• chronic administration• bioavailability and dosing route• elderly population- low pregnancy concern, compliance
The Pre-Clinical End Investigational New Drug (IND) application
• tox studies support dosing regimen (chronic tox)• bioavailability shows drug reaching target (BBB issue)• NCE is scalable
not necessarily not to scale
The Drug
The IND

IND ComponentsIND Application Table of Contents (from FDA form 1571)
1. Form FDA 1571 [21 CFR 312.23(a)(1)]
2. Table of Contents [21 CFR 312.23(a)(2)]
3. Introductory statement [21 CFR 312.23(a)(3)]
4. General Investigational plan [21 CFR 312.23(a)(3)]
5. Investigator’s brochure [21 CFR 312.23(a)(5)]
6. Protocol(s) [21 CFR 312.23(a)(6)]
a. Study protocol(s) [21 CFR 312.23(a)(6)]
b. Investigator data [21 CFR 312.23(a)(6)(iii)(b)] or completed Form(s) FDA 1572
c. Facilities data [21 CFR 312.23(a)(6)(iii)(b)] or completed Form(s) FDA 1572
d. Institutional Review Board data [21 CFR 312.23(a)(6)(iii)(b)] or completed Form(s) FDA 1572
7. Chemistry, manufacturing, and control data [21 CFR 312.23(a)(7)]
Environmental assessment or claim for exclusion [21 CFR 312.23(a)(7)(iv)(e)]
8. Pharmacology and toxicology data [21 CFR 312.23(a)(8)]
9. Previous human experience [21 CFR 312.23(a)(9)]
10. Additional information [21 CFR 312.23(a)(10)]

CMC- Chemistry, Manufacturing, ControlFrom Stuff to Drug Product

Chemistry, Manufacturing, ControlScaling up means more than making more
Discovery (Research)
• Maximize number of analogs• Salts and polymorphs not defined
Focus is on biological activity
• Low purity ≥ 90% Impurities usually not defined
• Stability unknown• Cost (relatively) unimportant
Working with milligram quantities
Development (Preclinical)
• Maximize yield/purity of 1-2 analogs• Salt form or polymorph defined
Focus is on scalability, stability, & formulation
• High purity ≥ 98%+ Impurities defined and qualified
≥ 0.05% must be reported≥ 0.1% must be qualified with Tox
studies
• Stability characterized• Cost of manufacturing / goods critical
Working with gram-kilogram quantities
Bench GMP

CMC - BiologicsCharacteristics of a good production cell line
• Grows rapidly - doubling times of 24-30 hrs
• Grows to high densities - > 107 cells/ml
• High production per cell - > 10 pg / cell / day
• Grows well in serum-free media - for ease of purification
• Genetically stable - protein production maintained for > 20 passages
• Recovery - high cell viability upon thawing of master cell bank
Getting to optimization:Oxygenation, CO2 level, pH, agitation, temperature, seeding density, split ratio, harvest timing
Common production cell lines:CHO, NSO, SP/2, BHK, Per.C

Chemical, Manufacturing, ControlConsiderations for biologics
• Monoclonal Antibodies– Various options for human or humanizing- with differing licensing fees and issues
• Production Cell Line– Different cell lines have different production levels- and licensing fees
• Master Cell Bank– A high yield, well characterized, stable, clonal source of cells producing the biologic
• Working Cell Bank– An aliquot from the master cell bank used for a production campaign
• Purification / Characterization– Sterility, mycoplasma, endotoxin, viral clearance

Current Good Manufacturing Process - cGMPSame is safer
• Requirements– Defined, written protocols– Complete records of all manufacturing and testing– Validation of key equipment and processes– Use of approved raw materials– Stability testing and controlled storage
• Release Specifications– Definition: the combination of physical, chemical, biological, and
microbiological test requirements that determine whether a drug product is suitable for release at the time of manufacture
– Examples: Identity Purity Physico-chemical properties (e.g. solubility, melting points, particle size, etc) Stability
– Requires validated analytical methods

Assay ValidationReproducible reproducibility
• Accuracy• Comparison of assay to existing measurements of analyte
• Precision• Agreement among individual test results (e.g. variance, standard deviation, coefficient of variation), including:
• Repeatability- intra-assay variability under same operating conditions (same operator, same day)• Intermediate Precision- intra-assay variability under varying conditions (different operator, different day)• Reproducibility- variability of assays performed in different laboratories
• Specificity• Performance of an assay in actual biological matrix (e.g. serum, saliva) and in presence of potentially interfering agents
• Detection Limit• Limit of detection (LOD) defined by spiking sample with a known quantity of analyte standard
• Quantification Limit• Lower limit of quantitation (LLOQ) and upper limit of quantitation (ULOQ) of analyte that can be measured accurately
• Linearity and Range• The range in which analyte concentration is proportional to assay readout
• Robustness• Effect of environmental variations (e.g. temperature, humidity) & performance variations ( e.g. incubation times)
Source: Guidance for Industry: Bioanalytical Method Validation, FDA CDER, May 2001

Analytical TestingEnd product: Certificate of Analysis (COA)
• Identity– Structure elucidation– IR, UV, NMR, MS, elemental analysis, wet chemical
• Strength– Measure of therapeutic activity– Assay, potency; HPLC, biological activity
• Quality– Measure of ingredient and manufacturing control– pH, optical rotation, specific gravity, viscosity, refractive index, dissolution,
disintegration
• Purity– Known impurities: intermediates, reagents/catalysts– Known constituents: moisture, organic volatile impurities, heavy metals,
arsenic, lead, sulfate, chloride– Biological: sterility, pyrogens, microorganisms
• Stability– Stress degradation study, define storage conditions– Typically 6 months to get to IND, eventually out to 2-3 years

Pharmacology and ToxicologySafety first
(Side effects may include…):•Eye Curdling
•Monkey Lung
•Brain Sporking
•Pituitary Frothing
•Follicular Swelling
•Pulmonary Weevils
•Fragile Hypothalamus
•Honus Wagner Disease
•Spontaneous Combustion
•Umbilical Cord Resurgence
•Tracheal Meerkat Colonies
•Chronic Temporal Inversion
•Sporadic Random Clinical Efficacy
*
* Thank you Stephen Colbert

Preclinical ToxicologyMoving from ADME to Clinic
• Objectives– Identify target organs
• CNS• Pulmonary• Cardiovascular
– Characterize dose response• NOAEL – No Observable Adverse Effect Level• MTD – Maximum Tolerated Dose
– Establish starting dose and escalation scheme– Determine reversibility of toxic effects– Stability testing and controlled storage
• Concerns– Narrow therapeutic index (LD50 / ED50)
– Irreversible– Delayed or cumulative

Definitive GLP Studies (GLP)IND-directed repeated dose toxicity studies
• Rodent and Non Rodent – Selected from range-finding studies
• Dosing Strategy– 4 dose groups: control (vehicle), test article (low, mid, high)– Route: generally by intended route & frequency
e.g. 3 times weekly for 4 weeks (for intermittent clinical administration) e.g. Daily oral dosing for 28 days (for daily clinical administration)
• Recovery Period– Usually ~2 weeks for 28 day study
• Analysis– Clinical Observations, Body Weight, Food Consumption– Clinical pathology (clinical chemistry, hematology, coagulation)– Urinalysis, Ophthalmology– Toxicokinetics (TK)
Determine drug profile and accumulation Day1 vs. Day 28– Histopathology (all tissues)– Identify MTD and No Observable Adverse Effect Level (NOAEL)

Genetic Toxicology (GLP)Generally required for all small molecules
• Bacterial Reversion Assay– Salmonella/E.coli Reverse Mutation Assay (Ames Test)
• In vitro mammalian cell assay– mouse lymphoma assay or– CHO chromosomal aberrations
• Micronucleus assay– May be performed as part of GLP rodent toxicity study
• Second tier (in vivo) test – may be required if a positive response in one of the above is
observed; options: Transgenic mutagenesis In vivo unscheduled DNA synthesis
• Biologics exception– Biologics generally do not require the in vitro tests, but
micronucleus may be requested as part of animal studies

Additional Safety TestingConsiderations for biologics
• Tissue cross-reactivity panel– Tests cross-binding of monoclonal antibody to non-target
tissues expressing the same or related epitope– Generally > 30 tissue types analyzed from multiple healthy
donors– Testing of binding to non-human tissues may no longer be
required
• Immunogenicity assay– The key issue is production of neutralizing antibodies– Generally, screening and confirmatory ELISAs are
followed by a functional assay to confirm effect beyond binding
– Example: sub-populations of MS patients who develop neutralizing antibody responses to beta-interferon

RegulatoryPreparing for the Spanish Inquisition

Regulatory ConsiderationsPreparing for the higher authority IND
• Pre Pre-IND Meeting- Informal scientific discussions between sponsor and FDA- Often used for tox questions
• Pre-IND Meeting (21 CFR 312.82)
- Common discussion topics:• Design of IND-enabling toxicology studies• Clinical protocol
- Submit questions in advance- Come with a plan- propose and listen- Submit a briefing document at least 4 weeks prior to a meeting- Identify the appropriate agency and contact
- (ODE1 Division of Neurology Products, Robbin Nighswander, (301) 796-1126)

FDA Guidance to IndustryNavigational help from your friends at the FDA
Oct 2006 Content and Formation of INDs for Phase 1 Studies of Drugs Including Well-Characterized, Therapeutic, Biotechnology-Derived Products
Jan 2006 Exploratory IND Studies
Jul 2008 Current Good Manufacturing Practice for Phase I Investigational Drugs
May 2009 Formal Meetings with Sponsors and Applicants for PDUFA Products
Dec 2010 Codevelopment of Two or More Unmarketed Investigational Drugs for Use in Combination (Draft Guidance)
Jul 2005 Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079645.pdf
CDER List of Guidances:

Clinical HoldWelcome to Hell
• Subjects exposed to unreasonable and significant risk of illness or injury
• Clinical investigators not qualified to conduct study
• Investigator brochure misleading, erroneous or materially incomplete
• Insufficient information submitted to assess risks to subjects

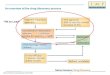
Project ManagementMore than getting to the train wreck on time…

A Guide to Preclinical DevelopmentHell’s roadmap
Background, rationale, and justification for
dose selection
Phase 1 Clinical Protocol
(protocol development)
Written pre-IND meeting request
Manufacture, control, and fill preclinical /clinical lot
Establish GMP manufacturing process, lot release
criteria, stability, uniformity
Finalize Phase 1clinical Protocol(s), ICFs
Certificate of Analysis, product characterization
Prepare CMC document
PrepareInvestigators’ Brochure
Pre-IND document
Finalize pre-clinical Toxicology protocol(s)
Pre-clinical toxicology testing in animals (GLP)
Final report(s)
Prepare integratedpharmacology/toxicology section
IND submission
FDA review and comment, Go / hold decision
Phase 1 Clinical Trials
pre-IND meeting & recommendations
PK/ Metabolism Dose range-findingFormulationAnalytical Methods Bioanalytical Methods
Non-GMP API
Pre-Clinical Toxicity Testing (protocol development)
Reg
Assay
CMC
ADME/Tox

Development Plan TimelineThe Gantt chart
Industry average = 3 years (18-24 months is reasonable)

Outsourcing OptionsWhen in Hell, it helps to have a Virgil
Grant and Contract Programs•ex: NIH RAID pilot program, Foundation Translational Programs
Contract Research Organizations (CROs)•CMC•ex: Aptech, Almac, Bachem, Bioreliance, Cambrex, Covance, Lonza
•Tox•ex: Absorption Systems, Apredica, BioTox Sciences, Charles River Labs, MPI Research, Southern Research, SRI International
CRO Directories•ex: Drug Discovery & Development (http://www.dddmag.com/article-2011-CRO-CMO-Directory-Preclinical-Testing-051311.aspx)•BioPharm International (http://www.biopharminternational.com/) •Biomedical Research Directory (http://www.biores.org/dir/Companies/Contract_Research_Organizations/)
What to look for / How to find it:•No violations (e.g. Form 483), experience of assigned project leader/team, security of IP, flexibility / problem solving, experience, availability / scheduling, reasonable price, quality of reporting•Network, consultants, program managers

Drug Development is a March of AttritionIntegrated Preclinical Development is part of the solution
CMC - It takes a scalable processSafety Toxicology - It takes multiple assessmentsProject Management - It takes a champion, it takes a team

AcknowledgementsIt takes a team
CMC• Alex Kostrikin [Anergen – PharmaCyclics]• Milan Tomic [Xoma]
Toxicology• Nancy Wehner [Anergen – Consultant]• Karen Steinmetz [SRI International]• Jon Mirsalis [SRI International]
Project Management• Tom Yonker [InterMune – KaloBios]• Joanne Snow [Snow Associates]